An example of the results obtained when labeling protocols and confocal acquisitions are carried out into the ischemic region is illustrated in Figures 1A and 1B. A two dimensional view of acquired images shows that at twenty-four hours after ischemia (A), the lysosomal marker CD68 (green) is expressed in hypertrophic ameboid CD11b cells (red) present in the ischemic core. In the border zone (B) CD11b positive cells display round cell bodies and ramified processes positive to CD68. Z-axis projections (right and bottom part of A and B) allow visualization of whether the marker distribution documented in the single plane view is also present along the z-axis. Where colocalization occurs, yellow pixels are generated. CD11b is the receptor for C3 fragments and exhibits a membrane distribution. CD68 labels lysosomes and is thus present mainly in the cytosol. CD11b/CD68 double positive cells usually have a small percentage of colocalized voxels and in most cases CD11b surrounds CD68 (Figure 1A). Only when phagosomes are bound to cell membrane during the phagocytic process, colocalization between CD11b and CD68 is realized (Figure 1B).
In Figure 2, the image processing that leads to the definition of marker coexpression is reported. In A, the three-dimensional view of confocal acquired images indicates the presence of CD68 positive cell (green) and Ym1 (red) positive cell, but it may not be used for the definition of colocalization. Indeed, fluorescent voxels laying parallel to the observer's view may yield a colocalized signal (yellow), that may not be related to the actual distance between markers (they are not solid objects and there may be transparency effects). To define colocalization, a single plane view with projections of the z-axis should be preferred (B). Moving along the Z-axis looking for colocalization (yellow pixels), the Z-axis projection is obtained (visible to the right and bottom part of B). Fluorescent voxels can be turn into solid objects by three-dimensional rendering (Figure 2C). The rotation of the volume allows the best view (from C' to C''''') for looking at the objects and properly assign marker expression to a specific cellular body. High magnification and solid view of three-dimensional renderings enable to define marker expression in clusters of cells (D-D''). After high magnification and 3D rendering, the two markers (CD68 and Ym1) appear to belong to different cells although in close proximity.
An example of the evaluation of the of M/M phenotype marker coexpression in the ischemic area is shown in Figure 3. Coexpression of CD11b (red), a marker of M/M activation/recruitment, CD68 (green) a marker associated with lysosomes as well as Ym1, expressed by alternative activated M/M are investigated in the ischemic area. To provide details on the functional status of M/M, we assessed their relationship with neurons (NeuN, in blue). Images of the sampled area (Figures 3A-3D) are evaluated as three-dimensional objects (Figures 3A'-3D'). CD11b/CD68 double positive cells are examined by three-dimensional rendering (Figures 3A' and 3B'). Analysis reveals that neurons (blue) are often enwrapped by CD11b positive cells in both ischemic core and border zone at 24 hr after ischemia. In most cases CD11b cells surrounding neurons are positive for CD68 at both zones, suggesting that these cells are engaged in active phagocytosis. None of the CD11b/Ym1 double positive cells (Figures 3C and 3C') appear to engage a phagocytic interaction with neurons, being CD11b/Ym1 double positive cells never in contact with NeuN positive cells.
| anti-CD11b | TSA amplification (Cy5) λ=646 nm | Alexa 546 anti-rat λ=532 nm |
| 1:800 | + | + |
| 1:1.500 | + | + |
| 1:3,000 | + | + |
| 1:5,000 | + | + |
| 1:10,000 | + | + |
| 1:30,000 | + | – |
| 1:40,000 | – | – |
Table 1. Fine tuning of working dilution for rat anti-CD11b antibody.

Figure 1. Coexpression of CD11b (red) and CD68 (green) 24 hr after pMCAO. Confocal microscopy for CD11b and CD68 shows that in the ischemic core CD11b cells are prevalently globular and some of them are positive to CD68 (A). In the border zone (B) both rounded and ramified CD11b cells are positive to CD68. Nuclei are in blue. Bars: 20 μm.

Figure 2. Coexpression of CD68 (green) and Ym1 (red) at 24 hr after pMCAO. Three-dimensional view of a confocal image acquired in the ischemic area, CD68 (green), Ym1 (red) and nuclei (blue) (A). Single plane view with z-axis projections excludes the presence of colocalized voxels (no yellow signal, B). Three-dimensional rendering turns fluorescent voxels into solid objects (C). The volume is rotated to find the best view (C'-C''''). Closer look to a cluster of cells (D). Three-dimensional rendering helps to define whether the markers are expressed by the same cell in the case of a cluster of cells (D'). CD68 and Ym1, although in close contact, are not coexpressed by the same cells, being clearly associated with different nuclei (D''). Bars: 5 μm. Click here to view larger figure.
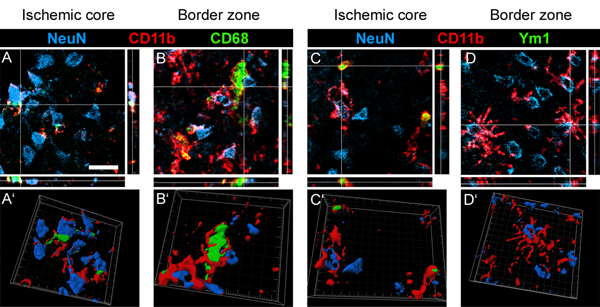
Figure 3. Coexpression of CD11b (red) and NeuN (blue) with CD68 (green) or with Ym1 at 24 hr after pMCAO. Confocal microscopy in the ischemic core (A; 3D rendering in A') and in border zone (B-B') reveals that CD11b/CD68 double positive cells envelop NeuN positive cells, possibly indicating phagocytosis of neurons. Ym1 positive cells are exclusively located into the ischemic core (C-D). Confocal analysis of CD11b/Ym1 double positive cells shows that they do not appear involved in a phagocytic interaction with neurons (NeuN positive cells, C; 3D rendering in C'). CD11b single positive cells in both ischemic core (C-C') and border zone (D-D') surround neurons. Bar: 20 μm. Click here to view larger figure.
