Electron cryo-tomograms of mitochondria clearly reveal the 3D morphology of the organelle (Figure 2). Manual segmentation of the membranes in a tomographic volume illustrates the structure of the cristae in a mitochondrion. By imaging mitochondria from different yeast knockout strains that lack certain protein components, the effect of these proteins on cristae morphology can by assessed. Figure 3 shows a mitochondrion from a yeast strain lacking ATP synthase subunit e. This component of the ATP synthase complex is required for the dimerization of the mitochondrial ATP synthase. Mitochondria from this strain lack the normal lamellar cristae of the wildtype mitochondria (Figure 2) and instead contain a number of inner membrane compartments. These compartments are either devoid of cristae or contain small balloon-shaped membrane invaginations (Figure 3).
In tomograms with good contrast, large mitochondrial protein complexes, in this case ATP synthase dimers, are easily visible (Figure 4; Movie 1). The structures of the complexes can be determined at 2-3 nm resolution by subtomogram averaging (Figure 5; Movie 2). The average volumes may be placed back into the tomogram in order to assess the organization of individual complexes relative to each other and to other protein complexes in the membrane (Figure 6; Movie 3).
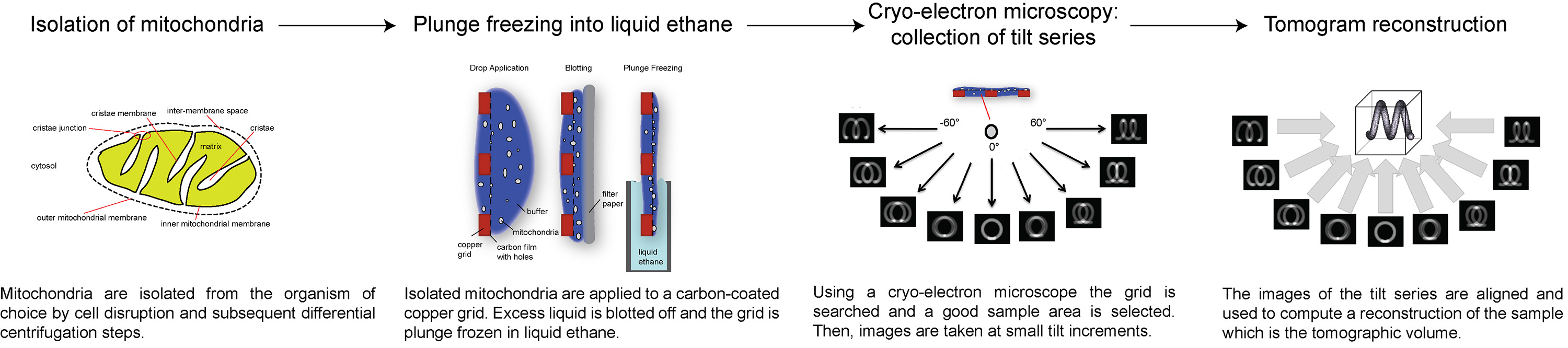
Figure 1. Flow-chart showing the stages of electron cryo-tomography . Click here to view larger image.
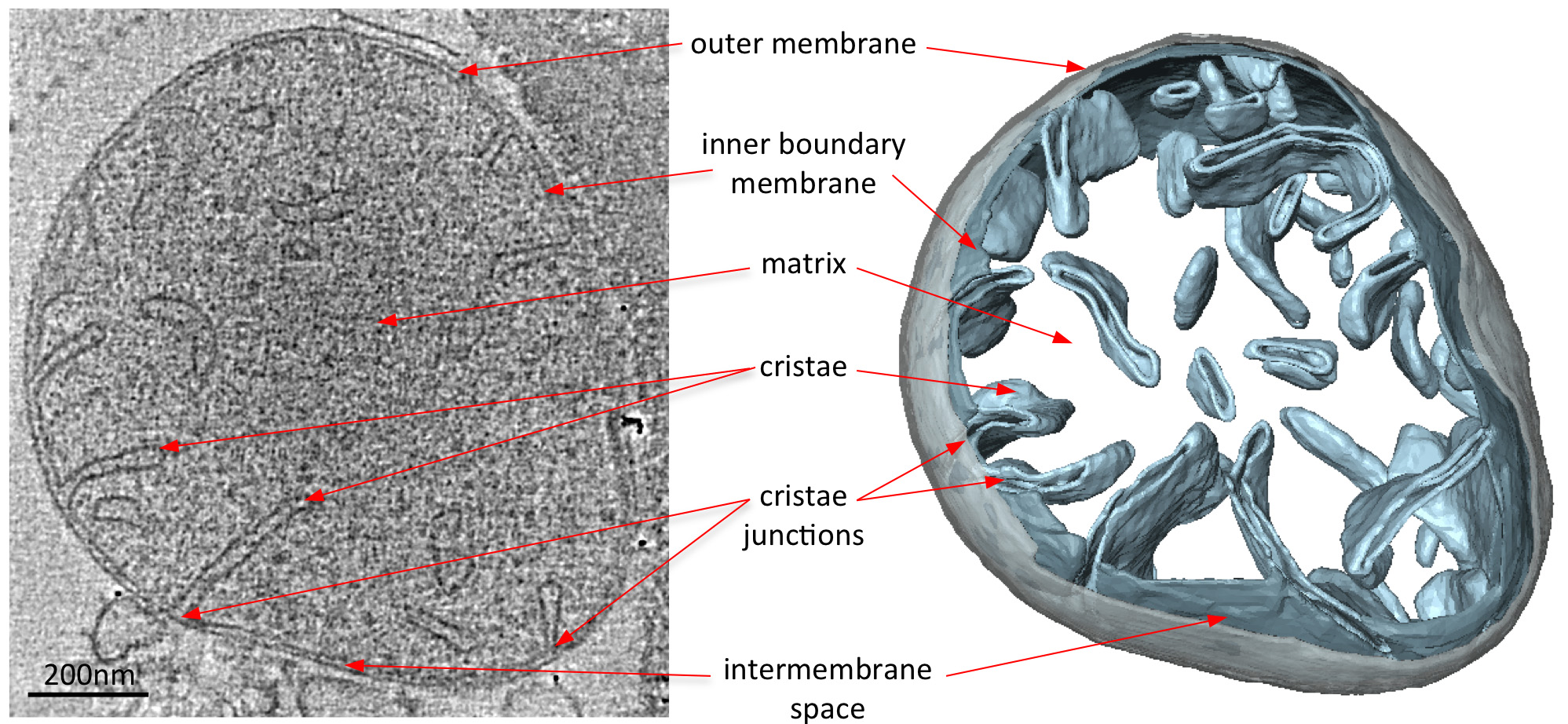
Figure 2. Morphology of a mitochondrion from wildtype S. cerevisiae. Central slice through a tomographic volume of a wildtype S. cerevisiae mitochondrion (left) and corresponding surface-rendered volume (right). The segmented volume of the outer membrane is shown in grey and the volumes of the inner boundary and cristae membranes in light blue. Adapted from Davies et al8. Click here to view larger image.
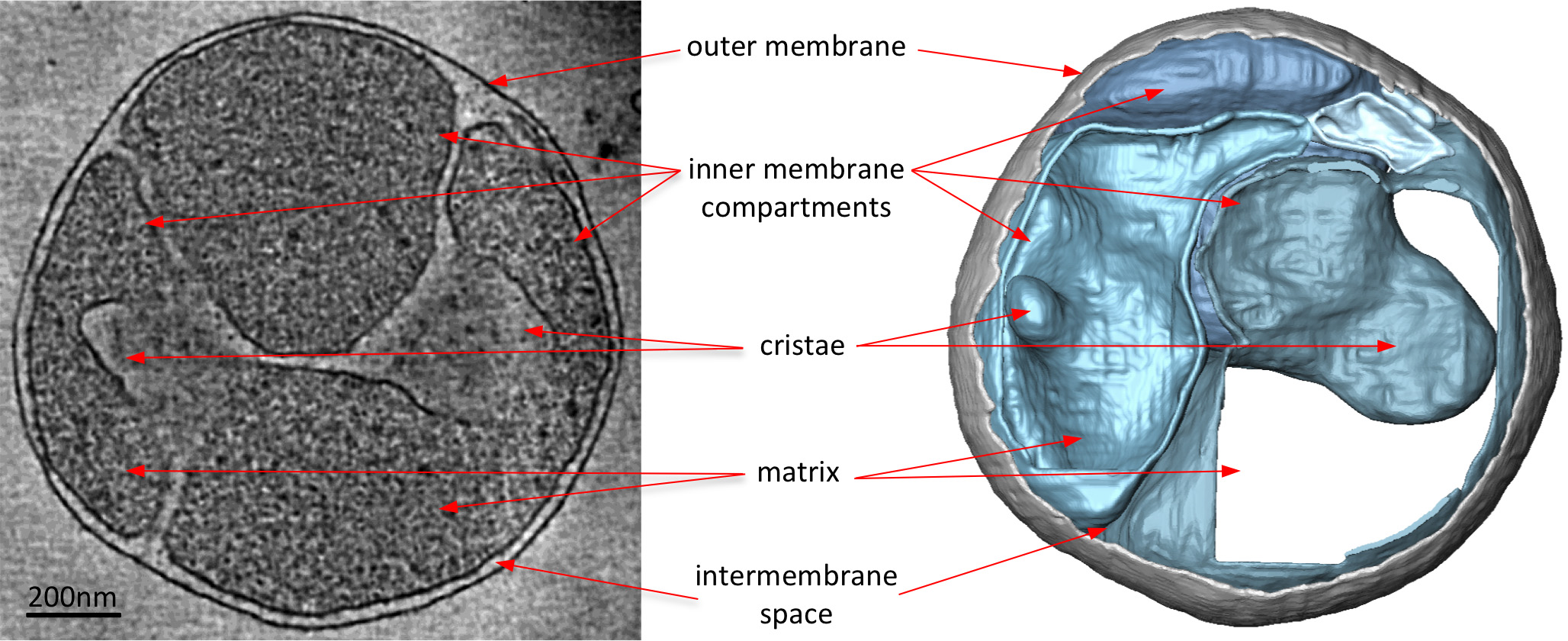
Figure 3. Mitochondrion from a S. cerevisiae strain lacking a subunit required for ATP synthase dimerization. Slice through tomographic volume (left) and accompanying surface-rendered volume (right) of a mitochondrion from a S. cerevisiae strain lacking the protein subunit e required for ATP synthase dimerization. When compared with Figure 2, the mitochondrion from the mutant strain lacks the normal lamellar cristae of wildtype mitochondria. Instead, the mitochondrion has many inner membrane compartments with either no cristae or balloon-shaped cristae. Thus electron cryo-tomography highlights alterations in membrane morphology due to gene deletions. Adapted from Davies et al8. Click here to view larger image.
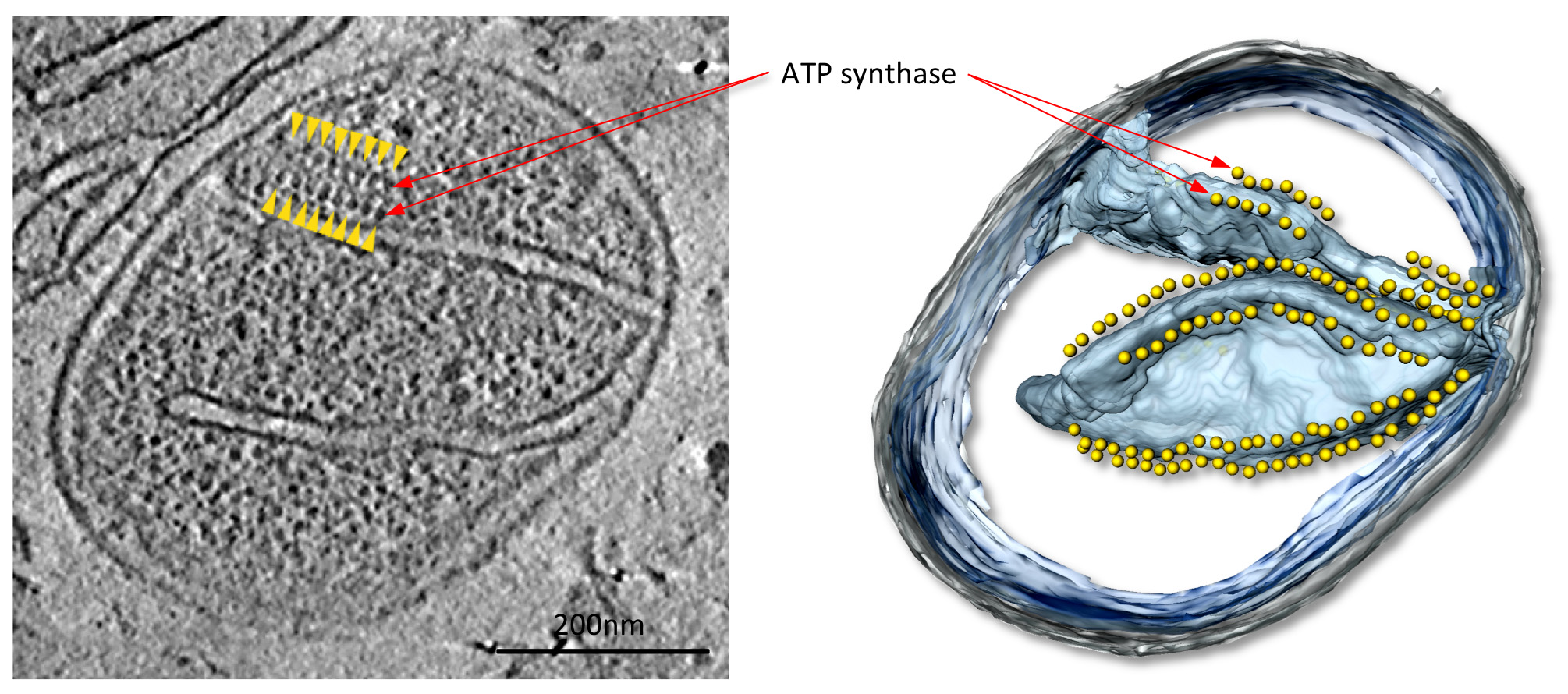
Figure 4. Mitochondrion from the fungus P. anserina. Slice through tomographic volume (left) and accompanying surface-rendered volume (right) of a mitochondrion from the filamentous fungus P. anserina. In this tomogram, rows of 10 nm particles (yellow arrowheads) are located above highly curved membrane ridges in the inner membrane cristae (see Movie 1). These particles were identified as ATP synthase dimers by subtomogram averaging. From Davies et al9. Click here to view larger image.
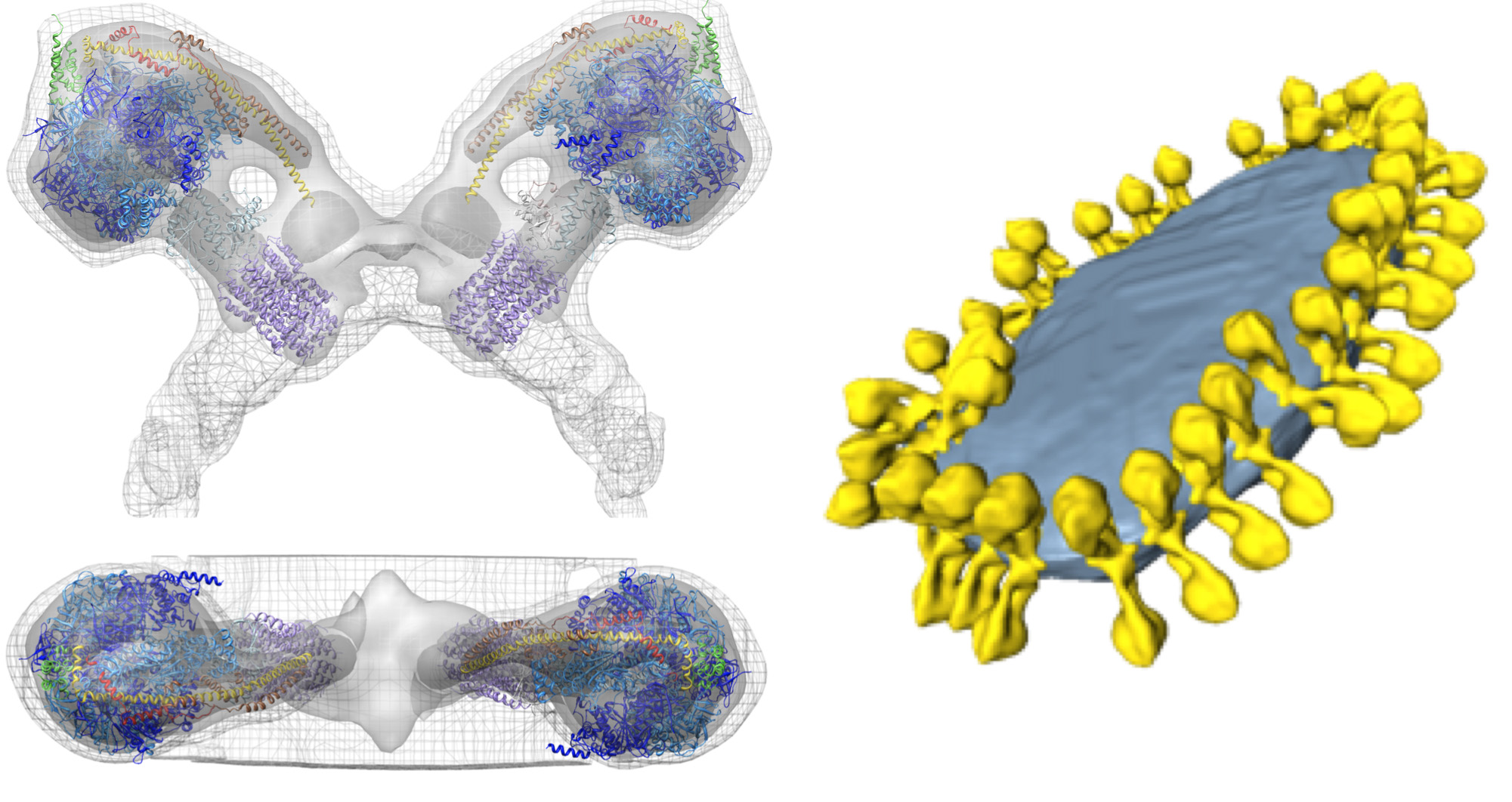
Figure 5. Structure and organization of the mitochondrial ATP synthase. Side and top view showing the electron density of an ATP synthase dimer from S. cerevisiae as determined by subtomogram averaging with fitted atomic models (left). Mitochondrial inner membrane vesicle showing the organization of ATP synthase dimers in rows (right). The figure was generated by positioning the subtomogram average of the ATP synthase dimer into the segmented volume of the membrane vesicle, using the coordinates calculated during averaging. Adapted from Davies et al8. Atomic models: F1/rotor-ring [PDB:2WPD]39 (blue and purple); oligomycin sensitive conferring protein-OSCP [PDB:2BO5]40 (green); peripheral stalk fragment [PDB:2CLY]1 with N-terminal residues from [PDB:2WSS]2 (yellow and red) (see Movie 2). Click here to view larger image.
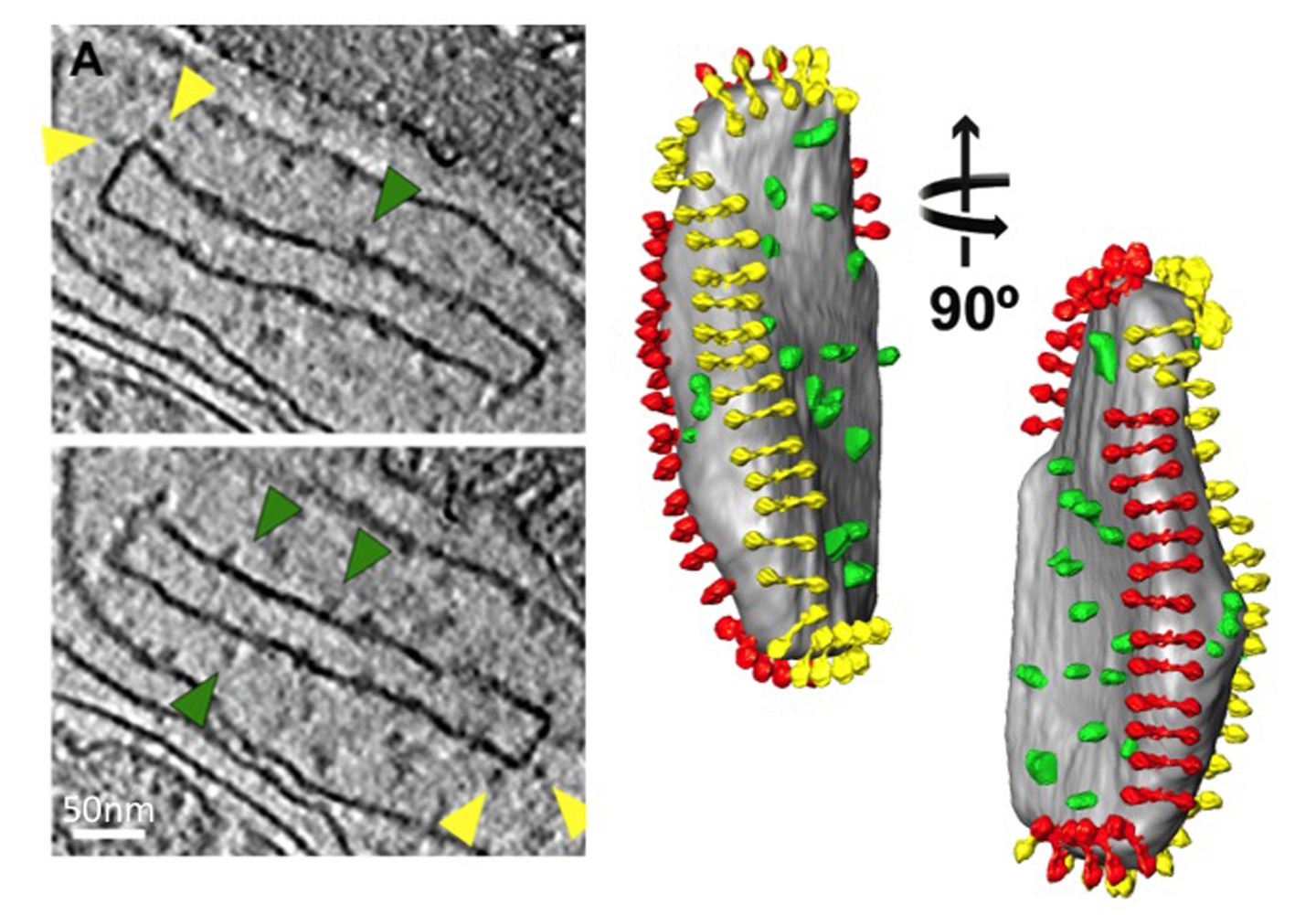
Figure 6. Isolated crista vesicle from a P. anserina mitochondrion. Slices from a tomographic volume (left) and accompanying surface-rendered volume (right) of a crista vesicle from P. anserina. Protein densities protruding from the membrane are clearly visible. The densities indicated by yellow arrowheads are ATP synthase dimers, as identified by subtomogram averaging. Green arrowheads point to densities identified by antibody labeling as NADH dehydrogenase (complex 1; for details see9). Segmentation of the protein densities reveals their organization in the cristae, with the ATP synthase dimers (red and yellow) forming rows along the highly curved cristae ridges and the NADH dehydrogenase complexes (green) in the membrane regions on either side of the rows (see Movie 3). From Davies et al9. Click here to view larger image.
Movie 1. Electron cryo-tomogram of a P. anserina mitochondrion. The movie shows successive slices through a tomographic volume taken of a mitochondrion from the filamentous fungus P. anserina. Rows of ATP synthase are indicated by yellow arrowheads. The surface-rendered, segmented volume shows the location of ATP synthase (yellow spheres) in relation to the 3D cristae structure. Outer membrane, grey; inner boundary membrane, transparent blue; cristae membranes, opaque blue. From Davies et al9. See also Figure 4. Click here to view video.
Movie 2. Subtomogram average of the mitochondrial ATP synthase dimer from S. cerevisiae with fitted atomic models. The average was calculated from 121 subvolumes. The density is displayed at three contour levels: 1s – mesh, 2s – light grey and 3s – dark grey. Atomic models were fitted into the density using the sequential fit routine in Chimera. Atomic models: F1/rotor-ring [PDB:2WPD]39 (blue and purple); oligomycin sensitive conferring protein-OSCP [PDB:2BO5]40 (green); peripheral stalk fragment [PDB:2CLY]1 with N-terminal residues from [PDB:2WSS]2 (yellow and red). Click here to view video.
Adapted from Davies et al8. See also Figure 5.
Movie 3. Isolated cristae vesicle from a P. anserina mitochondrion. Successive slices through the tomographic volume are shown, followed by the segmented surface-rendered volume. Protein densities protruding from the membrane are clearly visible. Red and yellow densities are ATP synthase dimers. Green densities are NADH dehydrogenase (complex I) as determined by antibody labeling. From Davies et al9. See also Figure 6. Click here to view video.
