

Summary
Ici nous rapportons le double étiquetage des cellules neurales de crête et des vaisseaux sanguins utilisant la greffe intraspecies de tube neuralde poussin GFP combinée avec l’injection intra-vasculaire de DiI. Cette technique expérimentale nous permet de visualiser et d’étudier simultanément le développement du système nerveux (entérique) dérivé de la CCN et du système vasculaire, au cours de l’organogenèse.
Abstract
Tous les organes en développement doivent être connectés à la fois au système nerveux (pour le contrôle sensoriel et moteur) ainsi qu’au système vasculaire (pour l’échange gazeux, l’apport en liquides et en nutriments). Par conséquent, les systèmes nerveux et vasculaire se développent côte à côte et partagent des similitudes frappantes dans leur architecture de ramification. Ici nous rapportons les manipulations embryonnaires qui nous permettent d’étudier le développement simultané du tissu nerveux crest-dérivé neural (dans ce cas le système nerveux entérique), et du système vasculaire. Ceci est réalisé en générant des chimères de poulet par l’intermédiaire de la transplantation des segments discrets du tube neural, et de la crête neurale associée, combinée avec l’injection vasculaire de DiI dans le même embryon. Notre méthode utilise des embryons transgéniquesde GFP de poussin pour le greffage intraspécifique, ce qui rend la technique de greffe plus puissante que le protocole de greffe interspécifique classique de caille-poussin utilisé avec beaucoup d’effet depuis les années 1970. ChickGFP- la greffe intraspécifique de poussin facilite l’imagerie des cellules transplantées et de leurs projections dans les tissus intacts, et élimine tout biais potentiel dans le développement cellulaire lié aux différences entre les espèces. Cette méthode tire pleinement parti de la facilité d’accès de l’embryon aviaire (par rapport à d’autres embryons de vertébrés) pour étudier le co-développement du système nerveux entérique et du système vasculaire.
Introduction
L’embryon de poulet est un organisme modèle inestimable dans la biologie du développement des vertébrés, notamment parce que son développement dans ovo permet des manipulations expérimentales qui sont autrement impossibles à effectuer chez les vertébrés qui se développent in utero. Cette accessibilité et cette facilité de manipulation ont permis à l’embryon de poussin de jouer un rôle clé dans de nombreuses découvertes marquantes dans le domaine de la biologie du développement. Parmi les techniques les plus puissantes a été l’utilisation d’embryons chimériques de caille-poussin pour étudier le destin cellulaire, une méthode mise au point par le professeur Nicole Le Douarin dans les années 19701-3. En particulier, les chimères caille-poussin ont été particulièrement utiles pour marquer et suivre génétiquement les populations de cellules de crête neurale (NCC) hautement migratoires au cours du développement précoce. Les NCC sont une population multipotente de cellules migratrices, apparaissant dans l’ectoderme dorsal aux marges du tube neural, qui donnent naissance à un large éventail de types de cellules dans tout l’embryon de vertébrés. Ceux-ci comprennent les structures craniofaciales (cartilage, os, muscles), les neurones et la glie (dans les systèmes nerveux sensoriel et autonome), les mélanocytes et une sous-population de cellules du système endocrinien2,4,5. L’un des facteurs les plus importants influençant le devenir des CNC est leur emplacement initial le long de l’axe antérieur-postérieur du tube neural. Par exemple, la NCC entérique, qui donne naissance aux neurones et à la glie du système nerveux entérique (ENS), provient de deux sous-populations discrètes: la première située dans la région vagale (cerveau postérieur caudal), et la seconde dans la région sacrée du tube neural6-13. Le greffage inter ou intra-espèces des régions correspondantes du tube neural ont été les techniques de choix pour étiqueter définitivement ces cellules et permettre par la suite le suivi, de leur naissance sur les marges du tube neural, jusqu’à leur destination finale dans le tube digestif6,7,10.
Une autre manipulation embryonnaire plus facile à effectuer chez le poussin, par rapport à d’autres modèles animaux, est l’étiquetage vital du système vasculaire. En effet, au fur et à mesure que l’embryon de poussin se développe, il repose sur un réseau vasculaire extra-embryonnaire qui fait circuler l’oxygène et les nutriments du jaune. Ce réseau vasculaire accessible, situé à la surface du jaune, peut être utilisé comme passerelle pour étiqueter le système vasculaire en développement de l’embryon au cours de l’organogenèse12,14-17. L’injection intravasculaire de divers colorants, tels que le colorant lipophile DiI, permet de délimiter/colorer tous les vaisseaux luminisés du réseau vasculaire naissant.
Parce que les organes en développement doivent être connectés à la fois au système nerveux (pour le contrôle sensoriel et moteur) ainsi qu’au système vasculaire (pour l’échange gazeux, l’apport de fluides et de nutriments), les deux réseaux se développent côte à côte et partagent des similitudes frappantes dans leur architecture de ramification18-20. Ici nous rapportons les manipulations embryonnaires qui nous permettent d’étudier le développement simultané de l’ENS NCC-dérivé, avec le système vasculaire, pendant l’organogenèse. Ceci est réalisé en générant des chimères de poulet par la transplantation de segments discrets du tube neural, y compris la crête neurale, combinée avec l’injection vasculaire de DiI. En tant qu’avancée par rapport aux chimères de poulet de caille, notre méthode utilise des embryons de poussins GFP transgéniques pour le greffage intraspécifique, ce qui rend la technique de transplantation plus puissante, en termes de cellules d’imagerie et de projections, et élimine tout biais potentiel lié aux différences entre les espèces.
Protocol
1. Préparation du micro-scalpel pour les ablations du tube neural
- Façonner un micro-scalpel à partir d’une aiguille à coudre en acier disponible dans le commerce.
- Aplatir d’abord l’aiguille des deux côtés à l’aide d’une meule montée sur une meuleuse de banc motorisée.
- Commencez à façonner le scalpel, d’abord sur une pierre de l’Arkansas de qualité grossière en utilisant un mouvement circulaire contrôlé, dans des directions alternées, des deux côtés de l’aiguille.
- Continuez les mêmes mouvements d’affûtage sur une pierre arkansas de qualité extra fine pour façonner un micro-scalpel ultra fin, avec un tranchant bien défini(Figure 1A, B).
REMARQUE: Les alternatives au micro-scalpel pourraient être des aiguilles électrolitiquement aigugées, des aiguilles de tungstène disponibles dans le commerce ou des aiguilles en verre tirées.
2. Incuber les œufs de type sauvage et de GFP jusqu’au stade souhaité
- Entreposer les œufs de poule fertilisés et les œufs de poule GFP transgéniques dans un incubateur refroidi à une température de 14 à 15 °C avant l’incubation, car le développement est arrêté à cette température. Conservez les œufs pendant quelques jours, jusqu’à une semaine.
- Pour commencer le développement, placez horizontalement les œufs de type sauvage et de GFP sur un plateau et incuber simultanément dans un incubateur à 37,5 ºC, de sorte que les embryons sont aux stades correspondants pour la greffe de tube neural.
- Pour obtenir des embryons au stade de développement de 10 à 12 somite pour effectuer une greffe de tube neural vagal, incuber des œufs pendant 1,5 jour (33 à 38 heures) et mettre au stade des embryons selon les tables de développement de Hamburger et Hamilton21.
3. Préparer les œufs pour le fenêtrage et la greffe
- Déplacez un œuf à la fois vers un porte-œufs sur mesure pour le fenêtrage. Faites un petit trou dans la coquille de l’œuf en tapotant à plusieurs reprises, avec des ciseaux droits, sur la surface supérieure de l’extrémité pointue de l’œuf.
- Retirer 2 à 3 ml d’albumine de l’œuf avec une aiguille hypodermique de 181/2 G et une seringue de 5 ml. Le retrait de l’albumine abaisse le jaune dans l’œuf et facilite le fenêtrage ultérieur sans causer de dommages à l’embryon.
- Jetez l’albumine. Scellez le trou avec une petite bande de ruban adhésif clair coupé à la taille avec de fins ciseaux.
- À l’aide de ciseaux incurvés, percez un autre trou dans la surface supérieure de la coquille d’œuf. Insérez la pointe des ciseaux dans le trou et, en gardant les ciseaux parallèles au banc, travaillez dans un mouvement circulaire pour couper une fenêtre d’environ 2 cm de diamètre sur le dessus de la coque.
- Gardez les ciseaux en position stationnaire et faites pivoter l’œuf. Jetez le disque retiré de la coquille d’œuf. À E1.5, l’embryon est reconnaissable comme un disque jaune plus foncé sur le jaune.
- Enlevez tout débris de coquille qui est tombé dans l’œuf à l’aide d’une pince à épiler. Jetez tous les œufs non fécondés (identifiés par une petite tache blanche sur le dessus sur le jaune clair).
4. Préparer l’embryon hôte à recevoir du tissu greffé
- Ajustez le stéréomicroscope au niveau des yeux et optimisez l’orientation de la source de lumière à col de cygne pour éclairer adéquatement l’embryon sans provoquer de réflexions.
- Pour visualiser l’embryon proprement dit,injectez une petite quantité d’encre de Chine sous le centre du disque jaune plus foncé, à l’aide d’un tube buccal et d’une micro-pipette en verre tiré(Figure 1C, Oii).
- Préparer l’encre 50:50 avec du PBS contenant de la pénicilline/streptomycine à une concentration finale de 100 μg/ml. Insérez la micro-pipette à travers la membrane jaune à l’extérieur du périmètre du blastoderme puis inclinez soigneusement sa pointe directement sous l’embryon.
- Délivrez de l’encre sous l’embryon en soufflant sur le tube buccal. Si le pipetage par la bouche n’est pas autorisé, utilisez plutôt une seringue de 1 ml. Veillez à ne pas introduire de bulles d’air sous l’embryon, ce qui peut entraîner une contamination, puis retirez soigneusement la micro-pipette en verre. Il s’agit d’une étape délicate qui peut entraîner la mort de l’embryon si elle n’est pas effectuée avec précision.
- Étagés l’embryon par référence à Hamburger et Hamilton21 et enregistrer l’étape dans un livre de laboratoire.
- À l’aide d’un micro-scalpel sur mesure (ou d’une aiguille de tungstène fine) monté sur un porte-aiguille, faites un très petit gaz dans la membrane vitelline, à côté de la zone où la micro-chirurgie sera effectuée.
- Appliquez soigneusement 2 à 3 gouttes de PBS sur la déchirure de la membrane (à l’aide d’une micro-pipette en verre et du tube buccal) pour créer de l’espace entre l’embryon et la membrane. Coupez une fenêtre plus grande dans la membrane pour exposer toute la région où la micro-chirurgie aura lieu.
- Retirez la région du tube neural d’intérêt à l’aide du micro-scalpel, en commençant par des incisions transversales rostrales et caudales sur l’ensemble du tube neural dorsal (au niveau de somite 1 à 7 dans la vidéo).
- Couper bilatéralement entre le tube neural et les somites pour séparer le tube neural des tissus environnants, sans endommager les somites.
- Séparez très doucement le tube neural du notochord sous-jacent, qui doit rester intact. Notez que l’excision réussie du tube neural laissera tous les tissus environnants parfaitement intacts (Figure 2).
- Retirez le tube neural excisé en l’aspirant dans une micro-pipette en verre, puis jetez-le.
- Notez le niveau d’ablation du tube neural dans un livre de laboratoire. L’embryon hôte est maintenant prêt à recevoir le tube neural du donneur.
5. Préparer le tissu greffé donneur
- Sélectionnez un embryon GFP fenêtré et apparié par scène en le visualisant sous un stéréomicroscope fluorescent avec filtre FITC. La fluorescence GFP permet de visualiser très facilement les somites et de mettre en scène l’embryon.
- Une fois qu’un embryon apparié au stade a été identifié, retirez l’embryon de l’œuf en faisant 4 incisions, avec des ciseaux à ressort Pascheff-Wolff (Figure 1C, l) dans une forme rectangulaire autour de l’embryon, puis ramassez-le doucement avec une cuillère à embryon.
- Placez l’embryon dans un verre de montre carré avec une base en polymère sylgard. Secouez doucement l’embryon avec du dumont #5 pince à épiler pour enlever tout jaune attaché. Retirez la membrane vitelline et épinglez l’embryon sur la base en polymère à l’aide de broches minutien en acier inoxydable(Figure 1C).
- À l’aide des ciseaux à ressort, faites 4 incisions de forme rectangulaire autour du tube neural et des somites environnantes, dans la même région qui a été retirée de l’embryon hôte.
- À l’aide d’une pipette de transfert en plastique, transférer le tube neural et les tissus somites de l’embryon GFP du donneur dans un verre de montre contenant 0,2% de pancréatine dans le PBS Pen/Strep.
- Laisser la digestion enzymatique se poursuivre pendant 10 min à TA pour aider à séparer les tissus. Après incubation en enzymes, utilisez des broches minutien en acier inoxydable montées sur une poignée pour séparer manuellement le tube neural de tous les tissus adjacents.
- À l’aide d’une micro-pipette en verre, transférer le tube neural dissocié vers un autre verre de montre contenant du DMEM + 10% de sérum(p. ex.chèvre, cheval ou veau fœtal) sur de la glace, pour rincer l’excès de pancréatine et arrêter la digestion enzymatique. Après 5 min, le tube neural disséqué est prêt à être greffé orthotopiquement dans l’hôte poussin(figure 2 et S1).
6. Greffer le tissu
- À l’aide d’une micro-pipette en verre, transférez soigneusement le tube neural disséqué du verre de la montre à l’embryon hôte. Placez le tube neural dans l’orientation antérieure-postérieure correcte et poussez doucement l’explant adjacent à la région excisée de l’hôte poussin à l’aide du micro-scalpel. Laissez un petit fragment d’ectoderme attaché à la surface dorsale ou en coupant une petite entaille pour identifier l’orientation du tube neural.
- Si nécessaire, utilisez le micro-scalpel pour couper l’explant à la taille exacte de la région excisée.
- Guidez doucement le tube neural dans la région ablated et positionnez-le de telle sorte que le côté dorsal soit correctement orienté. Utilisez une micro-pipette en verre, montée sur un tube buccal, pour enlever le PBS et/ou le liquide entourant le greffon. Cela aide le donneur et les tissus de l’hôte à adhérer et le greffon à s’établir.
- Scellez toute la fenêtre avec du ruban adhésif clair de 24 mm de large pour éviter la déshydratation et la contamination.
- Étiquetez l’embryon chimérique en le marquant avec un crayon sur la coquille d’œuf et notez son numéro dans le livre de laboratoire. Retournez l’œuf à l’incubateur pour un développement ultérieur.
7. Injecter diI dans les vaisseaux sanguins de l’embryon hôte
- Au moment expérimental souhaité (ici, 3 à 10 jours plus tard), récupérez l’embryon chimérique de l’incubateur et retirez le ruban clair à l’aide de ciseaux droits pour accéder à l’embryon dans l’œuf.
- Si nécessaire, agrandissez la fenêtre dans la coque à l’aide des ciseaux. Veillez à ne pas endommager la membrane chorioallantoïque si elle est fixée à la coquille, ce qui entraînerait une hémorragie et compromettrait l’étiquetage des vaisseaux sanguins.
- Choisissez une veine accessible sur le jaune en vous assurant que le flux sanguin est dirigé vers l’embryon. Choisissez un point de ramification de l’une des veines vitellines(figure 3B, C).
REMARQUE: À E6.5 - E7.5, la membrane chorioallantoïque peut devoir être doucement écartée avec une pince à épiler pour accéder aux veines jaunes. Après E8.5, la seule option est d’injecter dans l’une des veines membranaires chorioallantoïques puisque, à ce stade, la membrane chorioallantoïque recouvre entièrement l’embryon. - Retirez la membrane vitelline au-dessus du point d’injection choisi à l’aide de deux pinces Dumont #5 en la déchirant dans des directions opposées.
- Cassez une aiguille de verre tirée à l’aide d’un #5 Dumont et ajustez son diamètre à la taille approximative de la veine avant le chargement avec CellTracker CM-DiI. Faire la solution mère DiI à 40 μg/μl dans du DMSO et conserver à -20 °C. Préparer la solution de travail dans du saccharose/PBS 0,3 M à une concentration de 4 μg/μl.
- Aspirer entre 5 et 10 μl de DiI dans 0,3 M de saccharose/PBS dans l’aiguille à l’aide de l’aspiration avec un tube buccal. Les embryons plus âgés peuvent nécessiter jusqu’à 25 μl ou plus. À partir de E8.5, les embryons ont des veines plus grandes et plus musclées, qui peuvent avoir besoin d’être maintenues en position avec un #5 Dumont avant de poignarder avec l’aiguille de verre chargée de DiI.
- Insérez rapidement l’aiguille dans la veine et soufflez régulièrement avec le tube buccal pour permettre au DiI de rejoindre le flux sanguin lentement sans former de caillot. Vous pouvez également utiliser un injecteur de pression pour la livraison diI.
8. Prélever des embryons à des fins de sectionnement ou d’examen de montage entier
- Pour conserver autant de DiI dans l’embryon que possible, prélever l’embryon immédiatement après l’injection en le ramassant sur une cuillère perforée et en coupant les vaisseaux sanguins et les tissus conjonctifs avec une paire de ciseaux droits, pour libérer l’embryon du jaune.
- Retirez toutes les membranes lâches et disséquez les organes d’intérêt(c’est-à-direles poumons et le tube digestif dans ce tutoriel), en prenant grand soin de ne pas comprimer le tissu, ce qui crée une diffusion du DiI. Fixer immédiatement les tissus par immersion dans 4% PFA pendant 1 - 2 heures à RT.
- Rincer le tissu pendant 5 min dans du PBS, puis 15 min dans du PBS contenant 5 μg/ml de DAPI. Montez les échantillons sur une lame de microscope pontée pour l’examen de la monture entière ou intégrez-les pour la cryo-sectionnement.
Representative Results
La figure 1 montre les instruments typiques requis pour effectuer l’isolement microchirurgical et la transplantation du tube neural. La figure 2 montre la procédure de transplantation. Après la transplantation, les embryons sont examinés pour le succès de la transplantation. Cela implique d’examiner l’embryon sous un microscope à fluorescence stéréoscopique, généralement le lendemain de la microchirurgie, pour la présence de CNC dérivé du greffon (GFP +). Si la transplantation a été un succès, alors GFP + NCC peut être observé à proximité du tube neural et dans les voies de migration précoce menant vers le foregut. Si la procédure n’a pas réussi, GFP+ NCC ne sera pas observé à l’extérieur du tube neural, ou s’ils sont présents dans l’hôte, ils peuvent être en plus petit nombre. Ces embryons infructueux sont jetés. Typiquement, 5-8 greffes de tube neural sont effectuées en une journée, et de ces 80% sont réussies. Les raisons de l’échec de la transplantation du tube neural comprennent la mort de l’embryon en raison de lésions tissulaires subies pendant la microchirurgie, ou l’échec du tube neural à s’intégrer dans l’embryon hôte. Ce dernier peut résulter d’un mauvais placement du tube neural dans l’hôte ou d’un tube neural de mauvaise qualité dû à une mauvaise technique de dissection ou d’une exposition excessive à l’enzyme de dissociation. L’étape initiale de dépistage, ainsi que des examens ultérieurs similaires pour les cellules GFP +, est utile car elle signifie que le temps et les ressources ne sont pas gaspillés en effectuant des expériences sur des embryons qui n’ont pas de NCC marqué GFP dans l’intestin.
La figure 3 montre la procédure d’injection diI des vaisseaux sanguins. L’efficacité /succès de la technique d’injection DiI dépend de: premièrement, couper l’aiguille d’injection au diamètre optimal pour la veine ciblée, deuxièmement un geste précis lors de l’insertion de l’aiguille dans la veine (afin de ne pas percer de l’autre côté), et troisièmement éviter que l’aiguille se bouche pendant l’injection en soufflant à un rythme constant. Si l’un de ces trois paramètres est mal fait, l’embryon saignera ou aura besoin de plusieurs heures pour récupérer avant qu’une deuxième tentative ne soit faite car l’hémorragie rend presque impossible la réin injectation immédiate. Les embryons réussis doivent être sélectionnés immédiatement par visualisation au microscope à stéréofluorescence et doivent être disséqués rapidement. Dans les embryons réussis, les vaisseaux sanguins marqués diI sont présents dans tout l’embryon(figure 3C,D),y compris les lits capillaires(figure 3D).
Lors de la récolte d’embryons et de l’examen de sections de tissus ou de tractus gastro-intestinaux de montagne entière, les résultats typiques révèlent GFP + NCC dans l’ENS primitif et la structure fine des réseaux de vaisseaux sanguins intestinaux marqués DiI (Figure 4) Les préparations de montage entier peuvent être examinées à l’aide d’une microscopie confocale par laquelle les piles d’images produisent des reconstructions tridimensionnelles (3D) montrant les interrelations entre les projections fines des cellules GFP + ENS et le système vasculaire coloré diI (Figure 4 A-C; G-I; Vidéos 1 et 2).

Figure 1. Instruments de microchirurgie recommandés. (A) micro-scalpel en forme d’aiguille à coudre. (B) fine pierre de l’Arkansas pour façonner un micro-scalpel. (C) a) ciseaux droits, b) ciseaux incurvés, c) seringue de 5 ml avec aiguille hypodermique de 181/2 G, d) pipette en plastique, e) porte-œufs sur mesure, f) encre noire, g) verre de montre carré, h) verre de montre carré avec base sylgarde noire, i) micro-scalpel sur porte-aiguille, j) épingles minutien, k) aiguille minutien ou tungstène sur porte-aiguille, l) ciseaux à ressort Pascheff-Wolff, m) pince à épiler #5 Dumont, n) cuillère perforée, oi) aiguille de transfert courte tirée par le feu, oii) aiguille d’encrage longue tirée par le feu, p) tube buccal. Veuillez cliquer ici pour voir une version plus grande de cette figure.

Figure 2. Greffe intraspécifique de tube neural. Les images du tube neural de l’embryon de poussin/GFP ont été modifiées à partir de Delalande et al. 12.La vascularisation n’est pas nécessaire pour la colonisation intestinale par les cellules de la crête neurale entérique. Veuillez cliquer ici pour voir une version plus grande de cette figure.

Figure 3. Injection intraveineuse de DiI. (A) Instruments recommandés: a) CellTracker CM-DiI goutte sur parafilm, b) aiguille d’injection de verre tiré, c) tube buccal. (B) Schéma de schéma de l’injection intraveineuse de DiI dans l’embryon de poussin chimérique E4. (C) dans l’injection intraveineuse ovo DiI montrant l’aiguille de verre fine contenant DiI insérée dans la veine (flèche). (D) Embryon chimérique E4 après injection diI (rouge) avec tube neural GFP+ (flèche). (E) DiI a coloré le réseau de vaisseaux sanguins fins dans un embryon vivant, 24 heures après l’injection. Br: cerveau; H: cœur; LB: bourgeon de membre; R : allantois. Les images en (C) et (D) ont été modifiées à partir de Delalande et al. 12 ans La vascularisation n’est pas nécessaire pour la colonisation intestinale par les cellules de la crête neurale entérique. Veuillez cliquer ici pour voir une version plus grande de cette figure.

Figure 4 : Résultats représentatifs dans l’estomac et le caecum d’un embryon de poussin E5.5. (A-C) Reconstruction 3dimensionnelle (3D) d’une pile d’images confocales dans la région de l’estomac montrant (D) les cellules de la crête neurale entérique GFP+ (ENCC) (E) le système vasculaire coloré par DiI et (F) une image fusionnée des deux réseaux D-F Sections histologiques au niveau de l’estomac montrant (G) le GFP + ENCC (H) le système vasculaire coloré diI et (I) une image fusionnée des deux réseaux. Les noyaux sont colorés avec du DAPI (cyan). (G-H) Reconstruction 3D d’une pile d’images confocale dans la région caecum montrant (A) le front de migration GFP+ ENCC en vert, (B) le système vasculaire coloré diI en rouge, et (C) une image fusionnée des deux réseaux. Les images (A-F) ont été modifiées à partir de Delalande et al. 12 ans La vascularisation n’est pas nécessaire pour la colonisation intestinale par les cellules de la crête neurale entérique. Veuillez cliquer ici pour voir une version plus grande de cette figure.

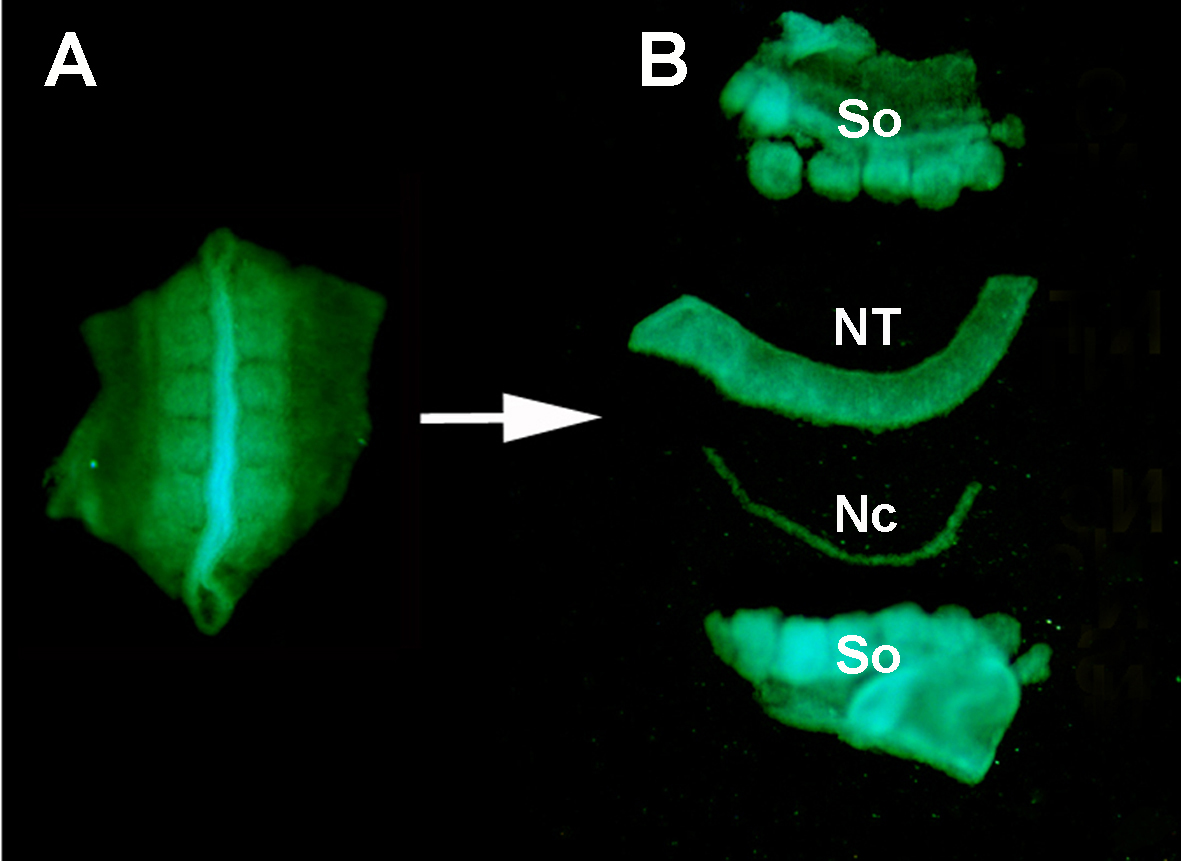
Figure S1. Isolement d’un tube neural GFP+ donneur des tissus environnants par digestion enzymatique et micro-dissection. (A) Tube neural GFP+ et somites adjacents disséqués de l’embryon de donneur. (B) Tube neural isolé après digestion de pancréatine et micro-dissection utilisant les broches inox minutien. Donc: somites; NT: tube neural; Nc: Notochord.

Vidéo 1. Rotation 3dimensionnelle à 360° de l’image sur la figure 4C, montrant le système vasculaire et l’ENCC dans l’estomac à E5.5 (HH27-28). Veuillez cliquer ici pour visionner cette vidéo.

Vidéo 2. Rotation 3dimensionnelle à 360° de l’image de la figure 4I, montrant le système vasculaire et le front de migration enCC dans la région du caecum à E5.5 (HH27-28). Veuillez cliquer ici pour visionner cette vidéo.
Discussion
La méthode de greffe intraspécifique du tube neural, combinée au étiquetage des vaisseaux sanguins décrit ici, tire pleinement parti de la facilité d’accès de l’embryon aviaire dans l’œuf (par rapport à d’autres embryons de vertébrés) pour étudier le co-développement d’un élément du système nerveux autonome (l’ENS) et du système vasculaire.
Pour l’étiquetage des dérivés ncc, la méthode de greffage intra-espèces de poussin gênéGFP-poussinque nous décrivons présente un certain nombre d’avantages par rapport à la méthode classique de chimère-poussin qui a été établie il y a plus de 40 ans1-3. Tout d’abord, sous la lumière FITC, la fluorescence GFP est extrêmement brillante, dans la mesure où les cellules GFP + sont facilement discernables dans les embryons chimériques vivants. Cela permet de vérifier le succès du greffon en ovo,alors que le greffage caille-poussin nécessite que l’embryon soit tué, traité et immunostained à l’aide de QCPN, avant que le succès du greffon puisse être constaté2. Deuxièmement, l’expression de la GFP chez le poussin transgéniqueGFP est cytoplasmique, elle marque donc non seulement les corps cellulaires, mais permet également de visualiser les projections des cellules transplantées22. Cela permet d’observer des réseaux neuronaux complexes à haute résolution (notez que les projections fines sont mieux visualisées lorsque l’échantillon est immunostained avec de l’anticorps anti-GFP). Comme l’étiquetage QCPN est limité au noyau cellulaire de caille, de tels réseaux ne sont pas révélés à l’aide de chimères de caille-poussin. Troisièmement, la greffe intraspécifique élimine toute différence potentielle entre les espèces au sein de l’embryon chimérique. Étant donné que les embryons de caille ont une période d’incubation plus courte que les poussins (19 jours contre 21 jours), il a été suggéré que les cellules de caille ont un taux de prolifération plus élevé que les cellules de poussin, ce qui pourrait potentiellement affecter le développement des tissus chimériques23. Fait intéressant également, il a été démontré chez les plantes que la greffe interspécifique pourrait produire des altérations importantes des modèles de méthylation de l’ADN chez l’hôte 24. Quatrièmement, laGFP des poussins facilite les expériences de rétro-transplantation pour aborder des sujets tels que le devenir des CNC et l’engagement cellulaire25. Cinquièmement, le poussin transgéniqueGFP est également utile pour de nombreuses autres techniques, notamment le tri FACS des sous-populations cellulaires GFP+, la culture organotypique d’organes contenant des cellules GFP+, la manipulation génétique des tissus greffés GFP+ par électroporation de plasmides d’expression26,et d’autres technologies d’imagerie telles que la tomographie par projection optique27.
L’approche de transplantation de tube neural peut être modifiée en remplaçant microchirurgical des quantités plus courtes de tube neural. En utilisant de plus petits segments du tube neural, la microchirurgie est potentiellement moins dommageable pour l’embryon et la survie peut être améliorée. Cependant, l’inconvénient de transplanter moins de tube neural est que le nombre de GFP + NCC dans l’hôte sera réduit. Les utilisateurs pourraient essayer d’atteindre un équilibre entre la quantité de tube neural transplanté pour donner une survie optimale des embryons, et le nombre de GFP + NCC dans l’intestin de l’hôte suffisant pour donner des résultats informatifs.
Pour la peinture de vaisseaux, DiI a l’avantage que sa fluorescence est très brillante et robuste. En outre, il a la capacité de diffuser lors de la fixation assurant la coloration des capillaires ouverts les plus fins. Comme il s’agit d’un colorant vital, les embryons peuvent survivre à la procédure d’injection et continuer à se développer avec un système vasculaire taché (jusqu’à 24 heures dans nos mains, bien que la coloration devienne plus ponctuée au fil du temps, voir la figure 3E). La combinaison de la greffe deGFP de poussin avec la peinture vasculaire de DiI est donc compatible avec l’imagerie en direct. Outre tous ces avantages, il est important de noter que l’injection vasculaire ne marque que les vaisseaux luminisés et n’identifie donc pas les capillaires non ouverts, les cellules endothéliales ou les cellules endothéliales isolées. Cependant, de nouveaux progrès dans la transgénèse aviaire pourraient fournir de nouveaux moyens de contourner ces problèmes, comme en témoignent les expériences utilisant des embryons de cailles Tg(tie1:H2B-eYFP) pour étudier la morphogenèse vasculaire28. Une autre limite de cette technique est que, pour un étiquetage efficace des vaisseaux dans les embryons à E7.5 et au-delà, de plus grandes quantités de colorant doivent être injectées, ce qui peut rendre les expériences coûteuses. Cependant, une modification de la technique pourrait inclure l’étiquetage des vaisseaux sanguins à faible coût à l’utilisation de l’encre surligneur14, bien que cette approche n’ait pas été essayée entre nos mains.
Les étapes critiques des procédures comprennent le processus de visualisation de l’embryon en injectant de l’encre sous le blastodisc. Si la membrane recouvrant le jaune est déchirée par l’aiguille remplie d’encre à ce stade, la survie de l’embryon est gravement compromise. En outre, il est important, lors de la préparation d’un tube neural donneur, que le tissu ne soit pas laissé pendant une période excessivement longue dans la pancréatine (considérez environ 10 min au maximum). Une exposition prolongée à la pancréatine endommage le tissu et le tube neural est alors difficile à manipuler et il ne s’intégrera pas bien dans l’hôte. L’acquisition d’une expérience de la technique d’injection diI sur des embryons de type sauvage est essentielle avant d’injecter des embryons chimériques, car une seule tentative d’injection est généralement possible pour chaque embryon. Le volume diI et le diamètre de l’aiguille sont des paramètres critiques pour chaque embryon et doivent être évalués sur le type sauvage, les contrôles de stade appariés.
En conclusion, notre double méthode d’étiquetage de la transplantation de tube neural et de la peinture de vaisseau de DiI dans des embryons vivants de poussin peut être employée pour étudier les inter-relations entre ncc et réseaux de vaisseau sanguin pendant l’organogenèse. Étant donné que les mécanismes responsables de l’établissement d’innervation et de vascularisation correctes de la cible au cours du développement des organes sont encore largement inconnus, cette méthodologie présente un potentiel pour de futures découvertes dans ce domaine.
Disclosures
Les auteurs déclarent qu’ils n’ont pas d’intérêts financiers concurrents.
Acknowledgments
Les œufs de poule GFP fécondés ont été fournis par le professeur Helen Sang, de l’Institut Roslin et de l’Université d’Édimbourg, au Royaume-Uni. L’installation de poulet transgénique Roslin est financée par le Wellcome Trust et par le Biotechnology and Biological Sciences Research Council (BBSRC). Le travail a été en partie financé, et soutenu par NT, par Great Ormond Street Hospital Children’s Charity, Londres, Royaume-Uni. Les auteurs remercient Ben Jevans, de l’Institut de la santé infantile de l’UCL, pour son aide dans la préparation d’embryons pour la greffe.
Materials
| Name | Company | Catalog Number | Comments |
| Fertilised chick eggs | Henry Stewart and Co, Louth, UK | ||
| Fertilised GFP chick eggs | The Transgenic Chicken Facility, The Roslin Institute, The University of Edinburgh | ||
| Egg incubator (Profi-H Hatcher) | Lyon Technologies, CA, USA | 910-033 | |
| 14C Incubator | Precision Cooled Incubator, Leec Ltd., Nottingham, UK | Model LT2 | |
| Stereo-microscope | LEICA | Model MZ 12.5 | |
| Digital Camera | LEICA | DC500 | |
| Image acquisition software | LEICA | IM50 | |
| Goose neck halogen cold light source | Advanced Imaging Concepts, Inc | KL 1500 LCD | |
| 181⁄2 G hypodermic needle | SIGMA - ALDRICH | HSWNH181 | |
| Pancreatin | SIGMA - ALDRICH | P3292 | |
| DMEM | SIGMA - ALDRICH | D5030 | |
| Goat serum | SIGMA - ALDRICH | G6767 | |
| 5 ml syringe | SIGMA - ALDRICH | Z248010 | |
| Mouth tube | SIGMA - ALDRICH | A5177 | |
| Sigma Pasteur pipettes non-plugged, L 5 3/4 in. | SIGMA - ALDRICH | S6018 | |
| Transfer pipettes, polyethylene | SIGMA - ALDRICH | Z350796 | |
| Borosillicate glass capillaries, thin wall without filament | Harvard apparatus | PY8 30-0035 | |
| Iris Scissors - ToughCut | Fine Science Tools | 14058-09 | |
| Curved Iris Scissors - ToughCut | Fine Science Tools | 14059-09 | |
| Needle holders (Nickel-plated pin holder) | Fine Science Tools | 26018-17 | |
| Pascheff-Wolff Spring Scissors | Fine Science Tools | 15371-92 | |
| Dumont #5 forceps | Fine Science Tools | 11251-30 | |
| Minutien pins | Fine Science Tools | 26002-15 | |
| Dumont AA forceps, Inox Epoxy- coated | Fine Science Tools | 11210-10 | |
| Perforated spoon | Fine Science Tools | 10370-18 | |
| Tungsten needles (0.125mm diameter) | Fine Science Tools | 10130-05 | |
| Sellotape (clear, 24 mm width) | Any Supplier | ||
| Pen/Strep (Penicillin, Streptomycin) Solution | VWR international | 101447-068 | |
| Sylgard 184 silicone elastomer kit | Dow Corning | S09 512 516 | |
| Pelikan black ink | Pelikan | 211-169 | |
| CellTracker CM-DiI | Molecular Probes | C-7001 | |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Molecular Probes | D1306 | |
| Settings for glass needle puller | Sutter Instruments | Flaming/Brown micropipette puller model P-86 | |
| Heat 950; Pull 150; Velocity 100; Time 200; Pressure 500 |
References
- Le Douarin, N. A biological cell labeling technique and its use in expermental embryology. Developmental Biology. 30, 217-222 (1973).
- Teillet, M. A., Ziller, C., Le Douarin, N. M. Quail-chick chimeras. Methods in Molecular Biology. 461, 337-350 (2008).
- Teillet, M. A., Ziller, C., Le Douarin, N. M. Quail-chick chimeras. Methods in Molecular Biology. 97, 305-318 (1999).
- Garcia-Castro, M., Bronner-Fraser, M. Induction and differentiation of the neural crest. Current Opinion In. Cell Biology. 11, 695-698 (1999).
- Bhatt, S., Diaz, R., Trainor, P. A. Signals and switches in Mammalian neural crest cell differentiation. Cold Spring Harbor Perspectives In Biology. 5, (2013).
- Burns, A. J., Douarin, N. M. The sacral neural crest contributes neurons and glia to the post-umbilical gut: spatiotemporal analysis of the development of the enteric nervous system. Development. 125, 4335-4347 (1998).
- Burns, A. J., Le Douarin, N. M. Enteric nervous system development: analysis of the selective developmental potentialities of vagal and sacral neural crest cells using quail-chick chimeras. The Anatomical Record. 262, 16-28 (2001).
- Burns, A. J., Delalande, J. M., Le Douarin, N. M. In ovo transplantation of enteric nervous system precursors from vagal to sacral neural crest results in extensive hindgut colonisation. Development. 129, 2785-2796 (2002).
- Burns, A. J., Champeval, D., Le Douarin, N. M. Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Developmental Biology. 219, 30-43 (1006).
- Wang, X., Chan, A. K., Sham, M. H., Burns, A. J., Chan, W. Y. Analysis of the sacral neural crest cell contribution to the hindgut enteric nervous system in the mouse embryo. Gastroenterology. 141, 992-1002 (2011).
- Goldstein, A. M., Hofstra, R. M., Burns, A. J. Building a brain in the gut: development of the enteric nervous system. Clinical Genetics. 83, 307-316 (1111).
- Delalande, J. M., et al. Vascularisation is not necessary for gut colonisation by enteric neural crest cells. Developmental Biology. 385, 220-229 (2014).
- Anderson, R. B., Stewart, A. L., Young, H. M. Phenotypes of neural-crest-derived cells in vagal and sacral pathways. Cell And Tissue Research. 323, 11-25 (2006).
- Takase, Y., Tadokoro, R., Takahashi, Y. Low cost labeling with highlighter ink efficiently visualizes developing blood vessels in avian and mouse embryos. Development, Growth & Differentiation. 55, 792-801 (2013).
- Bates, D., Taylor, G. I., Newgreen, D. F. The pattern of neurovascular development in the forelimb of the quail embryo. Developmental Biology. 249, 300-320 (2002).
- Mayes, P., Dicker, D., Liu, Y., El-Deiry, W. Noninvasive vascular imaging in fluorescent tumors using multispectral unmixing. BioTechniques. 45, 459-460 (2008).
- Li, Y., et al. Direct labeling and visualization of blood vessels with lipophilic carbocyanine dye DiI. Nature Protocols. 3, 1703-1708 (2008).
- Eichmann, A., Thomas, J. L. Molecular parallels between neural and vascular development. Cold Spring Harbor Perspectives In Medicine. 3, a006551 (2013).
- Weinstein, B. M. Vessels and nerves: marching to the same tune. Cell. 120, 299-302 (2005).
- Carmeliet, P., Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature. 436, 193-200 (2005).
- Hamburger, V., Hamilton, H. L. A series of normal stages in the development of the chick embryo. Journal of Morphology. 88, 49-92 (1951).
- Barraud, P., et al. Neural crest origin of olfactory ensheathing glia. Proceedings of the National Academy of Sciences of the United States of America. 107, 21040-21045 (2010).
- Senut, M. C., Alvarado-Mallart, R. M. Cytodifferentiation of quail tectal primordium transplanted homotopically into the chick embryo. Brain Research. 429, 187-205 (1987).
- Wu, R., et al. Inter-species grafting caused extensive and heritable alterations of DNA methylation in Solanaceae plants. PLoS One. 8, e61995 (2013).
- Freem, L. J., Delalande, J. M., Campbell, A. M., Thapar, N., Burns, A. J. Lack of organ specific commitment of vagal neural crest cell derivatives as shown by back-transplantation of GFP chicken tissues. The International Journal Of Developmental Biology. 56, 245-254 (2012).
- Delalande, J. M., et al. The receptor tyrosine kinase RET regulates hindgut colonization by sacral neural crest cells. Developmental Biology. 313, 279-292 (2008).
- Freem, L. J., et al. The intrinsic innervation of the lung is derived from neural crest cells as shown by optical projection tomography in Wnt1-Cre;YFP reporter mice. Journal of Anatomy. 217, 651-664 (2010).
- Sato, Y., et al. Dynamic analysis of vascular morphogenesis using transgenic quail embryos. PloS One. 5, e12674 (2010).

{kind=link}