

Summary
Biologia sintetica consente l'ingegneria delle proteine con proprietà senza precedenti tramite l'inserimento co-translational di aminoacidi non canonico. Qui, abbiamo presentato come una variante spettralmente rosso-spostato di un fluoroforo GFP-tipo con proprietà spettroscopiche di fluorescenza romanzo, chiamato proteina fluorescente "oro" (GdFP), viene prodotto in e. coli tramite incorporazione di pressione selettiva (SPI).
Abstract
Le proteine fluorescenti sono strumenti fondamentali per le scienze della vita, in particolare per la microscopia di fluorescenza delle cellule viventi. Mentre wild-type e ingegnerizzati varianti della proteina fluorescente verde da Aequorea victoria (avGFP) così come gli omologhi di altre specie già coprono gran parte dello spettro ottico, un gap spettrale rimane nella regione del vicino infrarosso, per quale fluorofori basati su avGFP non sono disponibili. Varianti della proteina fluorescente rosso-spostato (FP) sarebbero sostanzialmente espandere il toolkit per unmixing spettrale delle specie molecolari multiple, ma il FPs rosso-spostato naturali derivati da coralli o anemoni di mare hanno bassa resa quantica di fluorescenza e Foto-stabilità inferiore rispetto alle varianti avGFP. Ulteriori manipolazioni e possibile ampliamento del sistema coniugato di cromoforo verso la regione spettrale da' è limitata anche dal repertorio dei 20 amminoacidi canonici prescritto dal codice genetico. Per superare queste limitazioni, biologia sintetica può raggiungere ulteriori spettrale rosso-cambio mediante inserimento di aminoacidi non canonico nella triade cromoforo. Descriviamo l'applicazione di SPI a ingegnere avGFP varianti con nuove proprietà spettrali. Espressione della proteina viene eseguita in un triptofano-auxotrofi Escherichia coli ceppo e completando la crescita media con precursori di indole adatta. All'interno delle cellule, questi precursori vengono convertiti in analoghi corrispondenti triptofano e incorporati nelle proteine dalla macchina ribosomal in risposta ai codoni UGG. La sostituzione di Trp-66 nella variante "ciano" avanzata di avGFP (ECFP) di un elettrone-donante 4-aminotryptophan si traduce in GdFP con uno spostamento di Stokes 108 nm e una massima emissione fortemente rosso-spostato (574 nm), pur essendo termodinamicamente più stabile di suo predecessore ECFP. Incorporazione di specifici residui dell'amminoacido non canonico viene analizzato tramite spettrometria di massa. Le proprietà spettroscopiche di GdFP sono caratterizzate dalla spettroscopia di fluorescenza risolta nel tempo come una delle preziose applicazioni di FPs geneticamente codificato nelle scienze della vita.
Introduction
Dopo la scoperta della proteina fluorescente verde nella Medusa Aequorea victoria (avGFP) nel 19621 e la prima espressione eterologa in 19942 in altre cellule eucariotiche, sono diventate le proteine fluorescenti della famiglia GFP altamente utili strumenti e obiettivi nelle scienze della vita. Ingegneria genetica e molecolare vasta inclusa la regolazione dell'utilizzo di specie-codone, accelerazione di pieghevole, una migliore maturazione, una maggiore luminosità, prevenzione dell'oligomerizzazione e ottimizzazione delle proprietà spettrali e fotochimica compresa la possibilità di reversibilmente photoswitch3,4,5,6. GFP deve la sua fluorescenza da sua 4-(p- hydroxybenzylidene) imidazolidin-5-one (HBDI) cromoforo. Quest'ultimo autocatalytically è formato dalla triade cosiddetto cromoforo di aminoacidi (Ser-65/Tyr-66/Gly-67 in avGFP) dopo la formazione di un legame covalente aggiuntiva entro la spina dorsale del peptide sotto l'influenza di ossigeno molecolare7. Il sistema coniugato risonante stabilizzato interagisce dinamicamente con il suo ambiente molecolare, consentendo l'assorbimento nel campo visibile e fluorescenza verde caratteristica di queste proteine.
All'interno della triade di cromoforo, è obbligatoria la presenza di un aminoacido aromatico. Tuttavia, il repertorio standard dell'amminoacido comprende solo quattro residui aromatici (sua, Phe, Trp e Tyr). Questo limita la mutagenesi convenzionali approcci per raggiungere sostanzialmente più varianti di avGFP rosso-spostato rispetto il FPs naturale più rosso-spostato come DsRed8 da Discosoma striata coralimorphs o mKate/mNeptune9 da L'anemone di mare Entacmaea quadricolor. Di conseguenza, la parte da' e nel vicino infrarosso dello spettro ottico superiore a 600 nm è scarsamente coperto da varianti GFP. Questo è, naturalmente, una grave limitazione per approcci al microscopio di fluorescenza che richiedono demultiplexing spettrale delle diverse specie di fluoroforo allo stesso tempo. Ad esempio, indicatori di lunghezza d'onda sono anche necessari per fare uso del regime basso assorbimento del tessuto della pelle fra 700-1.000 nm nelle impostazioni per imaging10profondo del tessuto.
Fluorescente proteine derivate da avGFP sono suddivisi in diverse classi sulla base delle proprietà spettroscopiche e natura chimica dei loro cromofori11. Con la sua triade Ser-65/Tyr-Gly/66-67, il cromoforo di selvaggio-tipo esiste come un mix equilibrato tra la forma neutra, fenolico (λmax = 395 nm, ε = 21.000 M-1cm-1) e la forma anionica Fenolato (λmax = 475 nm, ε = 7.100 M -1cm-1), e lo spettro di emissione esibisce un singolo picco a 508 nm. Il gruppo dell'idrossile di Ser-65 è di importanza critica, come dona un H-legame al Glu-222 nelle vicinanze cromoforo (distanza: 3,7 Å), che promuove l'ionizzazione di questo carbossilato. Classe è caratterizzata da un cromoforo anionico Fenolato, come EGFP (64-Phe-Leu/65-Ser-Thr; λmax = 488 nm, ε = 35.600 M-1cm-1, λem = 509 nm). A causa della sostituzione di Ser-65-Thr(Ala,Gly), il picco di eccitazione 395 nm del modulo neutro fenolo è soppresso e il 470-475 nm picco della Fenolato anionico è cinque - sei volte avanzata e spostato a 490 nm. Classe II comprende proteine con un cromoforo fenolico neutro, come in zaffiro-GFP. Qui, la sostituzione di Thr-203-Ile quasi completamente sopprime l'eccitazione di 475 nm, lasciando solo il picco a 399 nm. Poiché il cromoforo anionico non può essere opportunamente solvatati, sua forma neutra è favorita. Classe III comprende le varianti fluorescenti "gialle" (EYFP; Ser-65-Gly/Val-68-Leu/ser-72-ala/THR-203-Tyr; Max ε λ = 514 nm, ε = 84, 600 M-1cm-1, λem = 527 nm) con interazione π-impilamento di una catena laterale aromatico e Fenolato, come realizzata dalle sostituzioni Thr-203-His(Trp,Phe,Tyr), che conducono a fino a 20 nm massimi di emissione rosso-spostato (Thr-203-Tyr). Ulteriore sostituzione (Gln-69-Lys) risultati in un altro 1-2 nm red shift a 529 nm, la variante di avGFP rosso-spostato più noto11. Lo scambio del fenolo per un indolo (Tyr-66-Trp) Crea classe IV, come la ECFP ciano-fluorescente (Ser-65-Thr/Tyr-66-Trp; λmax1 = 434 nm, ε = 24.800 M-1cm-1; λmax2 = 452 nm, ε = 23.600 M-1cm-1 ; Λem1 = 477 nm, λem2 = 504 nm). L'alloggio dell'indolo ingombrante è probabilmente abilitato da altri, mutazioni compensative. La maxima di eccitazione e di emissione di ECFP caduta inbetween quelli delle proteine con cromofori neutri o anionici. Proteine di classe V harbor un imidazolo al posto di fenolo (Tyr-66-His), ad es., proteine fluorescenti blu come EBFP. Classe VI è prodotto da un cambio di fenolo-a-Fenil favorendo la forma neutra cromoforo esclusivamente, che porta di conseguenza per le posizioni di picco eccitazione e di emissione più blu-spostato (360 nm e 442 nm, rispettivamente).
Mutagenesi sito-diretta classica è particolarmente adatta per la produzione di varianti di cromoforo romanzo avGFP, dalla permutazione del tripeptide e dei residui interagenti nella cornice di 20 aminoacidi canonici 65-67. Queste possibilità possono essere ulteriormente ampliate quando non canoniche varianti di aminoacidi aromatici vengono introdotti durante la sintesi di proteina ribosomale12. In linea di principio, ci sono due modi per eseguire questa operazione. La prima strategia si basa sulla tolleranza di substrato del macchinario traduzione della proteina, soprattutto della sintetasi del tRNA di aminoacyl (aaRSs) verso analoghi correlati dell'amminoacido. Per raggiungere questo obiettivo con alta efficienza, auxotrofico sforzi di espressione di Escherichia coli sono impiegati che non sono in grado di sintetizzare l'amminoacido naturale corrispondente. Questo permette la sostituzione di quest'ultimo con l'aggiunta di amminoacidi adatti non canonico (ncAAs) o precursori della stessa al terreno di coltura. Questa strategia, anche conosciuto come incorporazione di pressione selettiva (SPI)13,14, consente sostituzioni di residui specifici, che provocare globale incorporazione della ncAA. La seconda strategia utilizza codone di arresto tRNAs soppressore che pagano con la ncAA di engineered aaRS enzimi. Questo si traduce in readthrough di codoni di stop in-struttura e site-specific incorporazione di ncAA. Di conseguenza, questo metodo di soppressione di codone di arresto (SCS) conduce all'espansione del codice genetico15. Via mutagenesi, un codone di stop viene inserito nel gene target presso il sito desiderato. In linea di principio, SPI può anche essere utilizzato per creare peptidi ricombinanti e proteine che sopportano un'unica installazione di ncAA, dato che rari aminoacidi canonici come Met o Trp sono scelti per la sostituzione. Con Trp, SPI approcci hanno dimostrati di lavorare con una grande varietà di analoghi compreso 4 - F-, 5 - F - e 6-F-Trp, 7-aza-Trp, 4-OH - e 5-OH-Trp, nonché 4 - e 5-NH2- Trp o β (thienopyrrolyl) anche alanina derivati16 ,17,18,19,20. Così, SPI potrebbe essere molto vantaggioso per la sostituzione di aminoacidi aromatici di cromofori GFP di varianti non canonico per esplorare la possibilità di personalizzare ulteriormente gli spettri e shift di Stokes di questi FPs. Per quanto riguarda tutte le modifiche di sequenza della proteina, la compatibilità con maturazione pieghevole e cromoforo FP deve essere verificata sperimentalmente.
In questo lavoro, utilizziamo la classe IV ECFP21, che trasporta invece il selvaggio-tipo avGFP Tyr, Trp residuo all'interno sua triade cromoforo. Tramite SPI, questo Trp-66 (e Trp-57, il solo altri residui di Trp in ECFP) viene sostituito da 4-ammino-TRP. La presenza del gruppo amminico dell'elettrone-donante di 4-ammino-Trp entro il cromoforo favorisce la stabilizzazione di risonanza di un trasferimento di protone lontano rosso-spostato stato eccitato (ESPT) dotato di uno spostamento di Stokes 108 nm. Questa proteina fluorescente "oro" (GdFP) costituisce la variante con il più grande spostamento verso il rosso della fluorescenza massima (574 nm) tra tutte le proteine derivate da avGFP. Descriviamo il metodo di produzione della proteina GdFP di SPI e forniscono i protocolli per l'analisi obbligatoria delle proteine modificate risultante dalla spettroscopia di massa. Inoltre, mostriamo come GdFP può essere utilizzata e analizzata negli approcci di spettroscopia di fluorescenza risolta in tempo.
Protocol
1. trasformazione di Trp-auxotrofi Escherichia coli
- Trasformano chimicamente o electrocompetent cellule (50 µ l) di un Trp-auxotrofi Escherichia coli ceppo, ad es. ATTC 49980 (WP2, mutante derivata dal ceppo di e. coli B/R22), con 1 µ l di una soluzione acquosa di 1 ng / µ l del plasmide His6-ECFP pQE - 80L mediante shock termico o elettroporazione, rispettivamente. Consultare il Giove Science Education Database23,24 per i dettagli.
Nota: Il vettore di espressione His6 pQE - 80L-ECFP codifica un N-terminalmente 6 x His-Tag ECFP21 guidato da un promotore batterico T5 con operatore lac. Inoltre trasporta un marcatore di selezione AmpR e un colE1 origine di replicazione (la sequenza di spina dorsale pQE - 80L vettoriale può essere trovata su: https://www.qiagen.com/mx/resources/resourcedetail?id=c3b71572-4d82-4671-a79b-96357fe926d1&lang=en & autoSuggest = true). Il teorico peso molecolare della proteina di selvaggio-tipo His6-ECFG (dopo il cromoforo maturazione25) è Da 28303.92. Sequenza della proteina di destinazione convertito è come segue (His-tag sottolineato, sequenze di vettore-derivato in grassetto): MRGSALTHEOGSMVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTWGVQCFSRYPDHMK
QHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYISHNVYITADKQKNGIKANFKIRHNIEDGS
VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK. - Piastra di cellule trasformate su piastre di LB-agar (tabella 1) completate con glucosio 10 g/L, ampicillina di 100 µ g/mL e incubare le piastre a 37 ° C durante la notte.
2. espressione recombinant della proteina
-
Durante la notte cultura di e. coli ATCC 49980 pQE - 80L His6-ECFP
- Preparare 5 mL di terreno LB (tabella 1; completati con glucosio 10 g/L, ampicillina di 100 µ g/mL) in una sterile mL 14 cultura polistirolo tubo per crescita aerobica e inoculare con una colonia singolarmente da una piastra di agar utilizzando una pipetta sterile punta o inoculazione ciclo.
Nota: È consigliabile di utilizzare colonie dalle cellule appena trasformate. Le piastre con le colonie batteriche (dal punto 1.2.) possono essere memorizzate a 4 ° C per diversi giorni. - Incubare le cellule a 37 ° C in un agitatore orbitale a 200-250 giri/min per una notte.
- Preparare 5 mL di terreno LB (tabella 1; completati con glucosio 10 g/L, ampicillina di 100 µ g/mL) in una sterile mL 14 cultura polistirolo tubo per crescita aerobica e inoculare con una colonia singolarmente da una piastra di agar utilizzando una pipetta sterile punta o inoculazione ciclo.
-
Espressione di tipo selvatico ECFP
- Inoculare mezzo LB fresco 10 mL (tabella 1; completati con glucosio 10 g/L, ampicillina di 100 µ g/mL) con 100 µ l della coltura durante la notte in una beuta da 100 mL. Incubare la beuta a 37 ° C in un agitatore orbitale a 200 giri/min.
Nota: Facoltativamente, questo passaggio può essere eseguito in medium 10 mL NMM19 (tabella 1) completate con 100 µ g/L ampicillina e 0,5 mM L-triptofano (in alternativa, può essere utilizzato indolo). - Misurare la densità ottica a 600 nm (OD600) ogni 20 min. preferenzialmente misurare densità della cellula da determinare l'estinzione alle 600 nm (OD600) in uno spettrofotometro utilizzando una provetta con una lunghezza di percorso di 1 cm. sempre eseguire un riferimento misurazione utilizzando il terreno di coltura corrispondente. Diluire i campioni e mescolare i campioni bene per ottenere un valore di misura di 0.1-0.8, allora calcolare OD600 utilizzando il fattore di diluizione. Per informazioni dettagliate, fare riferimento al precedente pubblicazione 26.
- Dopo aver raggiunto un valore di600 OD 0.5-0.8 (circa 2-3 h dopo l'inoculazione), prendere il campione "prima di induzione" per SDS-PAGE (elettroforesi dodecilica del gel di poliacrilammide del solfato di sodio, passo 4).
- Indurre l'espressione della proteina bersaglio regolando la coltura liquida a 0,5 mM IPTG (isopropyl β-D-1-thiogalactopyranoside, dalla soluzione madre 1 M) e incubare a 30 ° C in un agitatore orbitale a 200 giri/min per 4-8 h.
Nota: Ciano proteine fluorescenti sono comunemente espressi a temperature inferiori a 37 ° C27. - Prendere esempio "dopo l'espressione" per SDS-PAGE (passaggio 4.).
- Raccogliere le cellule batteriche mediante centrifugazione per 10 min a 5.000 x g e a 4 ° C.
- Scartare il surnatante di decantazione e congelare il pellet di cellule a-20 ° C o da-80 ° C fino alla purificazione della proteina bersaglio.
- Inoculare mezzo LB fresco 10 mL (tabella 1; completati con glucosio 10 g/L, ampicillina di 100 µ g/mL) con 100 µ l della coltura durante la notte in una beuta da 100 mL. Incubare la beuta a 37 ° C in un agitatore orbitale a 200 giri/min.
-
SPI per la produzione di GdFP
- Inoculare in 10 mL di NMM19 medio (tabella 1) completati con 100 µ g/mL ampicillina, triptofano µM 15 e 10 µ l di cultura durante la notte in una beuta da 100 mL e incubare la beuta di cultura durante la notte a 30 ° C in un agitatore orbitale a 200 giri/min.
Nota: Una varietà di supporti chimicamente definite per la coltivazione di e. coli e SPI è disponibile. Oltre NMM utilizzati nel presente documento, MOPS medio28, glucosio-minerali sali medie29, Davis minimo medio30, M9 minimo medio31o GMML32 può essere utilizzato. - Il giorno successivo, misurare OD600 ogni 30 min fino a quando non cambia solo il valore di meno di 0.05 oltre 30 min. Il valore del plateau dovrebbe essere circa 1.
Nota: Deviazioni di ± 0,3 unità sono accettabili. A seconda del ceppo batterico e mezzo utilizzato, la concentrazione di triptofano iniziale (passo. 2.3.1) potrebbe essere necessario aggiustamento. - Prendere esempio "prima di induzione" per SDS-PAGE (passaggio 4.).
- Raccogliere le cellule batteriche mediante centrifugazione per 10 min a 5.000 x g e a 4 ° C. Scartare il surnatante di decantazione.
- Risospendere le cellule in 10 mL di terreno di NMM19 con ampicillina di 100 µ g/mL in una beuta da 100 mL e aggiungere 4-ammino-indolo a una concentrazione finale di 1 mM con soluzione di riserva di 50 mM. Continuare l'incubazione per 30 min a 30 ° C in un agitatore orbitale a 200 giri/min.
Nota: Questo passaggio è consigliato a causa della bassa stabilità chimica di ampicillina e assicura l'assorbimento cellulare di 4-ammino-indolo. - Indurre l'espressione della proteina bersaglio con l'aggiunta di IPTG ad una concentrazione finale di 0,5 mM mediante 1m stock e incubare il campione durante la notte a 30 ° C in un agitatore orbitale a 200 giri/min.
Nota: Ciano proteine fluorescenti sono comunemente espressi a temperature inferiori a 37 ° C27. - Il giorno successivo, misurare OD600.
- Prendere esempio "dopo l'espressione" per SDS-PAGE (passaggio 4.).
- Raccogliere le cellule batteriche mediante centrifugazione per 10 min a 5.000 x g e a 4 ° C ed eliminare il surnatante di decantazione.
- Nel caso in cui una nave non è stata utilizzata per centrifugazione, è possibile trasferire il pellet cellulare in una provetta di polistirene conica 50 mL con una spatola. Congelare il pellet cellulare a-20 ° C o da-80 ° C fino alla purificazione della proteina bersaglio.
- Inoculare in 10 mL di NMM19 medio (tabella 1) completati con 100 µ g/mL ampicillina, triptofano µM 15 e 10 µ l di cultura durante la notte in una beuta da 100 mL e incubare la beuta di cultura durante la notte a 30 ° C in un agitatore orbitale a 200 giri/min.
3. destinazione purificazione della proteina tramite cromatografia di affinità immobilizzata dello ione del metallo (IMAC)
-
Lisi della cellula batterica
- Scongelare il pellet cellulare in ghiaccio per 10-20 min.
- Risospendere il pellet cellulare in una provetta da 50 mL di polistirolo conico con 5 mL di buffer di associazione ghiacciata (tabella 1) sul ghiaccio.
- Aggiungere 20 µ l di lisozima di 50 mg/mL, 20 µ l di 1 mg/mL dnasi I e 20 µ l di 1 mg/mL RNasi r. Tappare la provetta, mescolare delicatamente capovolgendo 5 volte e tenerlo sul ghiaccio per 30 min.
Nota: Distruzione cellulare parziale si verifica come catalizzata dal lisozima. - Lisare le cellule di sonificazione usando una punta di omogeneizzatore ad ultrasuoni utilizzando tre cicli di 3 min in un tubo da 15 mL in polistirolo raffreddato da Granite con 2 s di impulso, 4 s di ampiezza di pausa e il 45%.
Nota: In alternativa, può essere utilizzato omogeneizzazione ad alta pressione, ad es., 20 cicli a 14.000 psi. Se necessario, diluire utilizzando buffer obbligatorio per raggiungere il volume minimo dello strumento. Inoltre, i reagenti di estrazione di proteine possono essere utilizzati per distruzione cellulare. Per esempi, vedere la tabella materiali. - Centrifugare il campione per 30 min a 15.000-18.000 x g, 4 ° C.
- Trasferire il surnatante in una nuova provetta e annotare il volume di liquido.
- Filtrare la soluzione attraverso un filtro per siringa da 0,45 µm utilizzando una siringa Luer lock plastica da 5 mL e un fluoruro di polivinilidene (PVDF) siringa filtro.
- Prendere esempio "lisato" per SDS-PAGE (passaggio 4.).
- Risospendere il pellet di detriti del cellulare in ddH2O (volume uguale come ex lisato).
- Prendere esempio "pellet" per SDS-PAGE (passaggio 4.).
-
Purificazione di IMAC
- Usare un 1 mL preimballati o self-imballato colonna IMAC FPLC (cromatografia liquida di proteine veloci) secondo le istruzioni del produttore. Utilizzare buffer obbligatorio (tabella 1) di equilibramento colonna anche per quanto riguarda la fase di lavaggio che segue dopo il lysate delle cellule è stato applicato alla colonna.
- Frazioni di eluato raccogliere e piscina con GdFP che possono essere identificati dal colore dorato chiaro visibile.
Nota: Facoltativamente, la proteina bersaglio può essere eluita utilizzando un gradiente lineare imidazolo (0-250 mM) utilizzando un sistema automatico di FPLC. - Determinare la concentrazione di proteina utilizzando il valore di letteratura per il coefficiente di estinzione presso 466 nm (ɛ466 nm = 23.700 M-1 cm-1)33 con tampone di eluizione come riferimento. Per informazioni dettagliate sulla procedura, consultare la precedente pubblicazione26.
- Prendere esempio "eluato" per SDS-PAGE e utilizzare 1-10 µ g di proteina per corsia in caso di Coomassie-macchiatura.
Nota: Gli importi di campione SDS possono variare a seconda del metodo di colorazione e la sensibilità di tintura. - Dializzare un'aliquota delle frazioni eluito contro dialisi buffer o MS buffer utilizzando una membrana con peso molecolare cutoff (MWCO) di 5.000-10.000. Preparare la membrana di dialisi secondo le istruzioni del produttore. Dializzare un campione di 1 mL tre volte contro 100 mL di tampone per almeno 2 h. Per ulteriori informazioni su questa procedura, fare riferimento alla precedente pubblicazione34.
- Per deposito, congelare il campione della proteina nel buffer di dialisi a-80 ° C.
Nota: Le aliquote devono essere stabile per almeno 6 mesi.
4. preparazione del campione SDS-PAGE di e. coli a cellula intera Estratto
- Trasferire una sospensione cellulare equivalente a 1 mL di OD600 = 1 sospensione (ad es. 500 µ l di OD600= 2) in una provetta da microcentrifuga da 1,5 mL.
- Raccogliere le cellule mediante centrifugazione per 10 min a 5000 x g, temperatura ambiente. Scartare il surnatante di pipettaggio.
- Aggiungere 80 µ l di ddH2O e 20 µ l di SDS 5x caricamento buffer di tintura (tabella 1) per il pellet cellulare e mescolare pipettando.
- Denaturare le cellule mediante riscaldamento a 95 ° C per 5 min in un blocco di calore o vasca di acqua. Successivamente, raffreddare i campioni a temperatura ambiente.
- Utilizzare 10 µ l per SDS-PAGE Coomassie tinto secondo precedente pubblicazione35.
Nota: Gli importi di campione SDS possono variare a seconda del metodo di colorazione e la sensibilità di tintura.
5. intatti proteina massa analisi mediante cromatografia liquida ad alte prestazioni (HPLC) accoppiata alla spettrometria di massa Electrospray ionizzazione Time-of-flight (LC-ESI-TOF-MS)
Nota: Buffer, impostazioni e HPLC gradiente può variare a seconda dello strumento utilizzato e la colonna di separazione. Cfr. la tabella materiali per attrezzature esemplare.
- Determinare la concentrazione di proteina da un campione dializzata contro MS buffer come descritto in precedenza (punto 3.2.3.) using MS buffer (Vedi tabella materiali) come riferimento.
- Diluire il campione di proteina a 0,1 mg/mL utilizzando MS buffer per un volume finale di 80 µ l, Miscelare pipettando attenta, trasferire la soluzione in un flaconcino di autocampionatore MS con inserto in vetro e chiudere con un tappo. Rimuovere le bolle d'aria, spostando il flaconcino.
- Riempire un secondo flaconcino di autocampionatore senza inserto in vetro (buffer vuoto) con 1 mL di tampone di MS.
- Consentire allo strumento per riscaldarsi. Eseguire la calibrazione dello strumento. Assicurarsi che una quantità sufficiente di solventi liquido cromatografia-grado è disponibile (> 100 mL).
- Programmare un gradiente HPLC lineare 20 min da 5% a 80% buffer un (0,1% acido formico in ddH2O), insieme con il tampone B (0,1% acido formico in acetonitrile).
- HPLC partono da un flusso di 0,3 mL/min e attendere che la pressione della colonna è stabile.
- Impostare un volume di iniezione campionatrice automatica di 5 µ l per il metodo di LC-ESI-TOF-MS, creare una lista di lavoro per un "bianco" seguito da un esempio di esecuzione e assegnare l'autocampionatore corrispondente flaconcino posizioni. Eseguire la lista di lavoro.
- Dopo il completamento della lista di lavoro, aprire il file di dati di esempio generati. Selezionare un intervallo nella trama (TIC) corrente ionica totale per la deconvoluzione e deconvolute lo spettro MS utilizzando l'algoritmo di deconvoluzione di massima entropia.
Nota: A seconda delle condizioni sperimentali, altre specie può verificarsi da FP non maturato o tampone ione addotti.
6. misure di durata fluorescenza e spettri di decadimento-collegata (DAS) di GdFP
Nota: Per la strumentazione di spettroscopia di fluorescenza risolta in tempo, fare riferimento alla Tabella materiali per attrezzature esemplare. Assorbanza, nonché l'eccitazione di fluorescenza e spettri di emissione delle proteine fluorescenti può anche essere registrati utilizzando spettrofotometri UV/Vis e fluorescenza di laboratorio.
-
Misurazione di vita di fluorescenza risolta in lunghezza d'onda di GdFP
- Preparare 2 mL di 1 soluzione µM di GdFP di diluizione in tampone PBS (tabella 1) a pH 7. Riempire la soluzione in una cuvetta di quarzo di 1 cm.
- Installare laser 470 ps-pulsata nm per l'eccitazione del campione e il filtro di emissione long-pass 488 nm e regolare la grata del fotone singolo lunghezza d'onda-correlati e nel tempo contando rivelatore36 (TWCSPC) per l'acquisizione del regime di lunghezza d'onda di 600 L/mm 500-700 nm.
- Acquisire l'emissione di fluorescenza ad un tasso di conteggio di circa 200 x 103 fotoni/s fino a circa 103 conteggi vengono accumulati in un massimo di acquisizione delle curve di decadimento della fluorescenza con single photon counting software.
-
Misurazione della risposta strumentale funzione37 (IRF)
- Sostituire la provetta del campione con una 1 cm di cuvette in quarzo riempita con silice colloidale di 1 g/L (~ 220 m2/g) in tampone PBS a pH 7.
Nota: La sospensione di silice viene preparata con una sospensione acquosa di 400 g/L. - Rimuovere 488 nm emissione long-pass e filtro inserto grigio per regolare il tasso di conteggio al rilevatore di TWCSPC inferiori a 100 x 103 conteggi/s.
- Regolare la griglia per l'acquisizione di fotoni a 470 nm a canale 8 del rivelatore TWCSPC 16 canali.
- Acquisire l'IRF fino a circa 10 x 103 conteggi vengono accumulati nell'emissione massima.
- Curve di decadimento fluorescenza Converti e IRF ai file di dati ASCII con global montaggio programma38 .
- Global di condotta adatta secondo un modello di una somma di tre componenti esponenziale con i corsi della vita come parametri collegati.
- Trama decadimento associato spettri (DAS) come distribuzioni di ampiezza dei componenti individuali di decadimento in dipendenza della lunghezza d'onda con software di analisi dati.
- Sostituire la provetta del campione con una 1 cm di cuvette in quarzo riempita con silice colloidale di 1 g/L (~ 220 m2/g) in tampone PBS a pH 7.
Representative Results
Utilizzando la tecnica dell'incorporazione di pressione selettiva, Trp-66 nella triade cromoforo ECFP (e Trp-57, il solo altri residui di Trp in ECFP) può essere sostituito da 4-ammino-Trp, generando in tal modo il GdFP rosso-spostato con proprietà spettrali distinte. Spettrometria di massa deve essere utilizzata per dimostrare l'integrazione stechiometrico desiderata dell'amminoacido non canonico nella proteina, con risultati mostrati nella Figura 1. In seguito, forniamo dati provenienti da microscopia, spettroscopia di assorbimento UV-Vis, nonché allo steady-state e risolto da tempo e lunghezza d'onda spettroscopia di fluorescenza per caratterizzare le proprietà del fluoroforo GdFP con un focus sulla dipendenza di pH della spettri.
Per confermare lo scambio di due residui di Trp in ECFP da 4-ammino-Trp, analisi spettrometria totale sono effettuato. La figura 1 Mostra uno spettro di ESI-MS deconvoluted rappresentanza di GdFP. Mentre il selvaggio-tipo ECFP ha una massa di proteine calcolato di 28,283.9 Da dopo la maturazione del cromoforo, la massa corrispondente di GdFP è Da 28,313.9. Lo spettro di ESI-MS deconvoluted di GdFP Mostra un picco di massa principale a 28,314.1 ± 0.1 Da, che si discosta dal valore teorico di meno di 10 ppm. Essendo all'interno della gamma di precisione tipica per questo tipo di analisi25, questo conferma l'incorporazione della ncAA tramite SPI (valore sperimentale per ECFP selvaggio-tipo: Da 28,283.7).
La figura 2 Mostra imaging microscopia in fluorescenza confocale immagini (CFIM) delle cellule batteriche che esprimono ECFP, EGFP, EYFP e GdFP su risospensione dei batteri in tampone PBS. Tutte le immagini sono state acquisite su un microscopio equipaggiato con un'eccitazione dei obiettivo e laser UV a circa la stessa energia per ogni campione.
Figura 3A Mostra una sovrapposizione di immagini CFIM di batteri e. coli che esprimono vari FPs tra cui GdFP, sempre monitorato con energia di eccitazione molto simile (lunghezze d'onda come in Figura 2). Figura 3B Mostra le strutture di cromoforo delle varianti FP mostrate. Per quanto riguarda la luminosità di GdFP rispetto al ECFP (fluorescenza quantistica resa φ = 0,4), EGFP (φ = 0,6) ed EYFP (φ = 0,6) è importante notare che per GdFP, una gamma più ampia di acquisizione della luce di fluorescenza (30 nm) è stato usato in contrasto con 20 nm utilizzato per tutte le altre spe CIES, al fine di regolare l'intensità delle immagini per valori simili. Con un coefficiente di estinzione leggermente più basso e un rendimento quantico ridotto in conseguenza di proprietà fotofisiche unico, la luminosità di GdFP è inferiore rispetto alla altri FPs mostrato.
Lo spettro di assorbimento di ECFP (Figura 3) ha due massimi caratteristici 434 nm e 452 nm. Al contrario, GdFP è caratterizzata da una banda di assorbimento di rosso-spostato ampio con il massimo a 466 nm. L'assorbimento di EGFP è ulteriormente rosso-spostato a 488 nm. Tuttavia, a causa dello spostamento di Stokes molto più grande di GdFP (108 nm) rispetto al ECFP (41 nm) ed EGFP (20 nm), lo spettro di emissione di GdFP è il più rosso-spostato di tutti i tre derivati GFP studiati qui (Figura 3D). Mentre l'emissione di fluorescenza di ECFP Mostra due massimi caratteristici a 475 nm e 505 nm, EGFP ha una vasta emissione principale band raggiunse la 508 nm (λmax) con una leggera spalla a 540 nm. La fluorescenza di GdFP appare a circa 565 nm (λmax.) (Figura 3D). Il suo spettro di emissione contiene un piccolo contributo di selvaggio-tipo ECFP che è anche visibile come una piccola spalla a 475 nm. Questa piccola frazione ECFP è sintetizzata prima induzione durante la procedura SPI, come descritto33.
3E figura Mostra le variazioni di pH-dipendente lo spettro di assorbimento di GdFP. Per un cambiamento di pH da 8 a 5, l'emissione massima si sposta leggermente verso il rosso e un lieve ampliamento della banda di assorbimento è osservato. Tuttavia, la riduzione dell'ampiezza di assorbimento è solo circa il 10% pH compreso fra 8 e 5, che indica che le proprietà di stato fondamentale del cromoforo GdFP sono molto debolmente per volta da pH.
Il tempo risolto l'emissione di fluorescenza monitorato dal conteggio di singolo fotone è illustrato nella Figura 4. Le curve di decadimento monitorate nei canali spettrali centrati a 550 nm e 600 nm (Figura 4A) Mostra un decadimento fluorescenza leggermente più veloce a 600 nm rispetto al deperimento a 550 nm. I risultati di una misura globale della fluorescenza decadono curve con due risultati esponenziali componenti in due componenti di decadimento fluorescenza spettralmente distinguibili con costanti di tempo di 1.0 ns e 3.3 ns (Figura 4 e D).
L'emissione di fluorescenza di GdFP fortemente dipende dal pH, come è tipico per molte varianti di proteina fluorescente della famiglia GFP. Figura 4B confronta l'emissione di fluorescenza di GdFP pH compreso fra 5 e 8, che mostra chiaramente una diminuzione dell'intensità di fluorescenza a basso pH, mentre le caratteristiche spettrali rimangono costante.
Il decadimento-collegata spettri (DAS)39 di GdFP (Figura 4 e D) sono caratterizzati da due bande di emissione distinta. Il contributo della 3.3 lento componente ns è più pronunciata nell'intervallo breve lunghezza d'onda intorno a 550 nm (60%) con un contributo minore del componente più veloce (40%). A 600 nm, entrambi i componenti hanno circa la stessa ampiezza. Al momento uno spostamento da pH 7 (Figura 4) a pH 6 (Figura 4), le caratteristiche spettrali del DAS difficilmente cambiare e le costanti di tempo dalla routine raccordo globale sono gli stessi (la precisione delle costanti di tempo DAS è di circa ± 0.15 ns). Tuttavia, la differenza tra le ampiezze assolute delle due componenti DAS è chiaramente evidente, che completamente rappresenta l'ampiezza di emissione di fluorescenza ridotta al momento lo stesso turno di pH in Figura 4B.
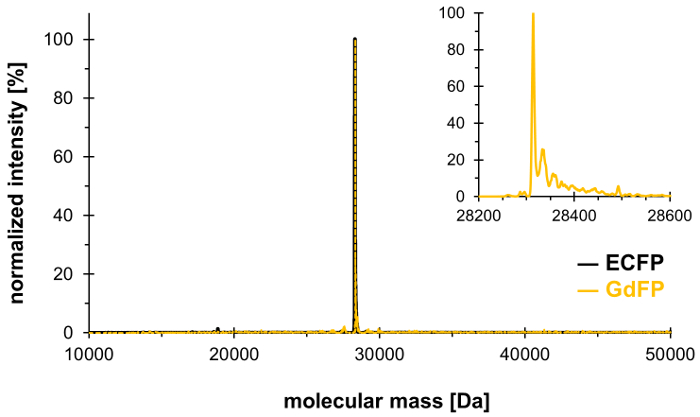
Figura 1: spettro di ESI-MS riconducibili rappresentativo di GdFP. Lo spettro di ESI-MS di GdFP (color oro, trama ingrandita mostrata come inserto) Mostra un picco principale presso 28314.1 Da (calcolato valore 28313.9 Da). Lo spettro di selvaggio-tipo ECFP è evidenziato in nero. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2: immagini di microscopia di fluorescenza confocale da popolazioni batteriche che esprimono vari FPs. Sono state utilizzate le seguenti impostazioni di lunghezza d'onda per acquisizione immagini: ECFP (λex = 457 nm, rilevamento: 461-480 nm), EGFP (λex = 488 nm, rilevamento: 495-515 nm), GDFP (λex = 476 nm, rilevamento: 560-590 nm), EYFP (λex = 514 nm, rilevamento: 520-530 nm). Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3: proprietà spettrali di GdFP. (A) CFIM immagine di una miscela di cellule batteriche esprimendo ECFP, EGFP e GdFP dopo la risospensione dei batteri in tampone PBS. (B) strutture di cromoforo di GdFP (con 4-ammino-Trp al posto di residui 66), la ECFP parentale (con Trp alla posizione 66) ed EFGP (con Tyr posizione 66). (C) confronto tra gli spettri di assorbimento normalizzati di GdFP, ECFP ed EGFP, considerando che in (D), lo spettro di emissione di fluorescenza normalizzata di ECFP (eccitazione a 430 nm) viene confrontato con lo spettro di emissione di fluorescenza di EGFP e GdFP (entrambi eccitato a 450 nm). (E) pH-dipendenza degli spettri di assorbimento (normalizzato di assorbimento a 280 nm). Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4: fluorescenza risolta in tempo di GdFP. Fluorescenza (A) deperimento del GdFP monitorati da tempo e lunghezza d'onda-risolto singolo fotone conteggio nei canali spettrali centrati a 550 nm e 600 nm (± 12,5 nm) dopo l'eccitazione con gli impulsi laser di 470 nm. La funzione di risposta strumentale (IRF) fornisce informazioni sulla risoluzione di tempo del programma di installazione utilizzato. (B) variazione dello spettro di emissione di GdFP dipende dal pH (eccitazione a 460 nm). (C, D) Spettri decadimento-collegata (DAS) di GdFP a pH 7 (C) e pH 6 (D) determinato dopo deconvoluzione di fluorescenza risolta da tempo e lunghezza d'onda decade e raccordo globale dei decadimenti in tutti i canali di un insieme globale di due funzioni esponenziali con costanti di tempo collegato. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 5: strutture di trasferimento della carica intramolecolare di ECFP (nero) e GdFP (oro) cromofori. L'aumento della dimensione del sistema di un gruppo amminico come parte della ncAA cromoforo del donatore di elettrone buona consente la formazione di strutture più mesomere per raggiungere la stabilizzazione di risonanza dello stato eccitato. I punti di connessione all'impalcatura FP sono mostrati come semicerchi. Clicca qui per visualizzare una versione più grande di questa figura.
| Soluzione di riserva | concentrazione, solvente | Nota | |
| 20% D-glucosio | 200 g/L D-glucosio in ddH2O | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| indolo | 50 mM in isopropanolo | ||
| 4-ammino-indolo | 50 mM in etanolo al 20% (20 mL di etanolo in un volume finale di 100ml riempito con ddH2O) | ||
| IPTG | DdH2O di 1 M | ||
| L-triptofano | 15 mM disciolto in ddH2O utilizzando 1 M HCl (aggiungere goccia a goccia HCl sotto agitazione fino a quando la polvere è dissoved) | ||
| lisozima | 50 mg/mL in ddH2O | ||
| Dnasi I | 1 mg/mL in ddH2O | ||
| RNasi A | 1 mg/mL in ddH2O | ||
| Amp100 | ampicillina 100 mg/mL in ddH2O | ||
| sodio-dodecilsolfato (SDS) | 200 g/L in ddH2O | ||
| solfato di ammonio ((NH4)2SO4) | DdH2O di 1 M | sterilizzare in autoclave | |
| potassio fosfato monobasico (KH2PO4) | DdH2O di 1 M | sterilizzare in autoclave | |
| idrogeno fosfato di potassio (K2HPO4) | DdH2O di 1 M | sterilizzare in autoclave | |
| solfato di magnesio (MgSO4) | DdH2O di 1 M | sterilizzare in autoclave | |
| D-glucosio | DdH2O di 1 M | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| cloruro di sodio (NaCl) | DdH2O di 5 M | sterilizzare in autoclave | |
| cloruro di calcio (CaCl2) | 1 g/L | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| cloruro ferroso (FeCl2) | 1 g/L | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| tiamina | 10 g/L | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| biotina | 10 g/L | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| mix di oligoelementi | solfato di rame (CuSO4), zinco cloruro (ZnCl2), il cloruro di manganese (MnCl2), molibdato di ammonio (NH4)2MoO4; ogni 1 mg/L in ddH2O | sterilizzare mediante filtrazione attraverso un filtro di siringa dimensione dei pori da 0,45 µm | |
| 19 aminoacidi mix | 1.) sciogliere 0,5 g di L-fenilalanina e 0,5 g di L-tirosina in 100 ml ddH2O con aggiunta goccia a goccia di 1 M HCl sotto agitazione fino a quando la polvere si è sciolta. | ||
| 2.) pesa 0,5 g di ciascuno dei rimanenti L-amminoacidi (ad eccezione di L-triptofano). Mescolare con 22 mL fo 1m KH2PO4 e 48 mL di 1 M K2HPO4. Aggiungere ddH2O a circa 800 mL. Mescolare fino a quando la soluzione diventa chiara. | |||
| 3.) Aggiungi il disciolta L-fenilalanina e L-tirosina dal punto 1.) e regolare il volume a 1 L con ddH2O. | |||
| 4.) sterilizzare la miscela dell'aminoacido di filtrazione sotto vuoto con un'unità di filtro superiore della bottiglia. | |||
| Buffer e Media | Composizione/preparazione | ||
| SDS caricamento buffer di tintura, 5x concentrato | 0.25 M Tris pH 6.8, 50% v/v glicerolo, 0.25% w/v bromofenolo Blu, didhiothreitol 0,5 M (DTT; in alternativa 5% β-mercaptoetanolo), 10% w/v sodio-dodecilsolfato (SDS) | ||
| buffer di associazione | 50 mM sodio dihydrogenphosphate (NaH2PO4), 500 mM NaCl, imidazolo di 10 mM, pH 8 | ||
| tampone di eluizione | 50 mM sodio dihydrogenphosphate (NaH2PO4), 500 mM NaCl, imidazolo 250 mM, pH 8 | ||
| buffer di dialisi | 50 mM sodio dihydrogenphosphate (NaH2PO4), 150 mM NaCl, glicerolo 100 mL/L, pH 8 | ||
| Buffer di MS | 10 mM Tris-HCl, pH 8 | ||
| nuovo terreno minimo contenente 19 L-amminoacidi tranne L-triptofano (NMM19) | Mescolare tutte le soluzioni di riserva per ottenere le concentrazioni finali seguenti: 7,5 mM (NH4)2SO4, 1.7 mM NaCl, 22mm KH2PO4, 50mm K2HPO4, 1mm MgSO4, 20 mM D-glucosio, 50 mg/L di 19 aminoacidi mescolare, 1 µ g/L CaCl2, 1 µ g/L FeCl2, 10 µ g/L tiamina, biotina 10 mg/L, mix di oligoelementi di 0,01 mg/L | ||
| LB medium | Composizione: tryptone di 10 g/L, Estratto di lievito 5 g/L, NaCl, 10 g/L pH 7.0 in ddH2O | ||
| Preparazione: | |||
| 1.) pesare fuori tryptone 50 g, 25 g di lievito estratto, 5 g di NaCl in una bottiglia di vetro 1L. | |||
| 2.) Aggiungi ddH2O fino a ~ 800 mL e componenti di sciogliere sotto agitazione. | |||
| 3.) misurare il pH e regolare a pH 7 di aggiunta goccia a goccia di 1 M HCl o NaOH M 1, se necessario. Aggiungere ddH2O fino a 1 L. | |||
| 4.) sterilizzare in autoclave, controllare in seguito per la perdita di volume e aggiungere ddH sterile2O per compensare se necessario. Conservare a 4 ° C fino all'utilizzo. | |||
| Piastre di agar LB | Composizione: tryptone di 10 g/L, Estratto di lievito 5 g/L, 10 g/L NaCl, agar-agar 15 g/L, pH 7.0 in ddH2O | ||
| Preparazione: | |||
| 1.) pesare fuori tryptone 50 g, 25 g di lievito estratto, 5 g di NaCl, agar-agar 7,5 g in una bottiglia di vetro 1L. | |||
| 2.) Aggiungi ddH2O fino a 500 mL e si dissolvono componenti sotto agitazione. | |||
| 3.) misurare il pH e regolare a pH 7 di aggiunta goccia a goccia di 1 M HCl o NaOH M 1, se necessario. Aggiungere ddH2O fino a 1 L. | |||
| 4.) sterilizzare in autoclave, controllare in seguito per la perdita di volume e aggiungere ddH sterile2O per compensare, se necessario. (Nota: LB agar può essere memorizzato a 4 ° C fino all'utilizzo per la preparazione di piatti di agar della libbra. Con cura si fondono agar solidificato utilizzando un forno a microonde) | |||
| 5.) quando la soluzione è ancora calda (30-40 ° C), aggiungere ampicillina a una concentrazione finale di 100 µ g/mL | |||
| 6.) pour circa 15 mL di liquido dal punto 5.) in un sterile 10cm di Petri in condizioni sterili. Quando l'agar si è solidificato, piastre possono essere archiviati per 1 settimana a 4 ° C fino all'utilizzo. | |||
| tampone fosfato salino (PBS) | Composizione: 137 mM NaCl, 2.7 mM KCl, 10mm Na2HPO4, 1.8 mM KH2PO4, 1mm CaCl2, 0,5 mM MgCl2, pH 7. Sterilizzare in autoclave o filtrazione. | ||
Tabella 1: Soluzione Stock e buffer.
Discussion
Per ottenere rendimenti molto elevati di incorporazione ncAA, il metodo SPI Auxotrofia basato si basa sull'uso di cellule metabolicamente derivati dal host, che non sono in grado di sintetizzare la controparte naturale corrispondente della ncAA. Per e. coli, tali ceppi sono facilmente disponibili. Anche l'incorporazione simultanea di più ncAAs nella stessa proteina è fattibile usando ceppi di multiauxotrophic. La modalità di specifici residui di sostituzione e il repertorio chimico essendo limitato agli analoghi chimici simili può essere visto come svantaggi. Tuttavia, un gran numero di varianti della proteina può essere prodotto come l'apparato di traduzione batterica naturale tollera numerose dell'amminoacido analoghi. Ad esempio, più di 50 ncAAs potrebbe essere incorporato nelle proteine usando la traduzione in vitro , pari a circa il 73% di tutti i codoni del codice genetico di essere disponibili per la riassegnazione40. Inoltre, SPI può anche permettere di etichettatura multisito efficiente della proteina bersaglio41. In linea di principio, la metodologia SPI non è limitata ad e. coli, ma possa lavorare in qualsiasi altro host e per ciascuno dei 20 aminoacidi canonici, purché auxotrofi ceppi e definita di coltura media è disponibili. Ad esempio, due analoghi di metionina, azidohomoalanine (Aha) e homopropargylglycine (Hpg), sono commercialmente disponibili e utilizzati per l'etichettatura di proteine e proteomi in diversi organismi. Inoltre, Aha può essere prodotta intracellulare e successivamente incorporò nella proteina42. Questa ncAA è particolarmente adatto per bioorthogonal coniugazioni come chimica clicca come sviluppato da Tirrel e collaboratori: ad esempio, nel tessuto di Arabidopsis thaliana, Bombyx mori larve43, Drosophila della pianta cellule44, zebrafish larvale45 così come le cellule di mammiferi tra cui neuroni46, proteine possono essere etichettate con Aha47,48. Allo stesso modo, Trp analoghi con successo furono incorporati in peptidi antimicrobici in Trp-auxotrofi Lactococcus lactis ceppi49. SPI è anche utile per il campo di Xenobiologia50,51, che Esplora alternative per la composizione chimica di base della vita. Ad esempio, basato su precedenti lavori su52 di e. colie Bacillus subtilis53, un ceppo di e. coli è stato sviluppato recentemente da una strategia evolutiva con pressione selettiva di utilizzare thienopyrrole invece di indolo, con conseguente sostituzione di tutto il proteoma di triptofano da thienopyrrole-alanina nel codice genetico54. Generalmente, l'aminoacido canonico Trp, che è codificato da una sola tripletta (UGG), presenta un bersaglio promettente per l'ingegneria delle proteine a causa il ricche sfaccettature della chimica di indolo, che offre numerose variazioni chimiche. Recentemente e come alternativa all'incorporazione di SPI-basato, un romanzo SCS piattaforma in grado di incorporare Trp analoghi site-specifically in sia batteriche che eucariotiche è stato segnalato55. Questo amplia ulteriormente la casella degli strumenti di in vivo l'ingegneria delle proteine basato su ncAA, tra cui l'alterazione delle proprietà spettrali.
Oltre all'utilizzo di host espressione auxotrofi, il protocollo SPI richiede condizioni di fermentazione rigoroso, sia in termini di tempi di espressione di destinazione e la composizione del mezzo al fine di raggiungere l'alta efficienza di incorporazione di ncAA e obiettivo di rendimento di proteina 56. coltivazione è condotta usando media minimi chimicamente definite, che essenzialmente contenga oltre sali principali fonti per azoto (sale di ammonio) e carbonio (D-glucosio), vitamine e oligoelementi. Benché non sia obbligatorio in assenza di ulteriori auxotrophies, i rimanenti aminoacidi (20 -n, se n aminoacidi vengono sostituiti) sono comunemente aggiunti per promuovere la crescita batterica57. Durante una fase di crescita iniziale prima dell'induzione dell'espressione della proteina bersaglio, gli aminoacidi canonica n essere sostituito vengono aggiunti nel limitare le concentrazioni. Crescita cellulare procede finché non gli aminoacidi essenziali mirati sono vuotati, indicato come sperimentalmente da un fisso OD600. Successivamente, il terreno di coltura viene sostituito da mezzo fresco che manca dell'aminoacido impoverita e contiene la ncAA in concentrazioni abbondanti. Per l'incorporazione di ribosomal di analoghi di triptofano come mostrato in questo protocollo, un indolo analogico è alimentato, che diventa intracellulare convertiti il corrispondente derivato di triptofano dal triptofano sintasi58. Successivamente, viene indotta l'espressione della proteina bersaglio. In questa fase, le cellule sono vicine alla fine della crescita logaritmica, come un equilibrio tra il numero totale delle cellule e il fitness. Come la presenza e l'incorporazione di Canonica amminico porterebbe alla produzione di proteina wild-type, è fondamentale per garantire che l'amminoacido essenziale è completamente esaurito prima dell'induzione. Allo stesso modo, è obbligatorio per esaminare l'efficacia dell'incorporazione di ncAA nel proteina bersaglio, comunemente mediante spettrometria di massa. In caso di sostanza presenza di canonica dell'aminoacido, la coltivazione condizioni c'è bisogno di essere regolata, per esempio, modificando la concentrazione dei amino acid(s) essenziali per la fase iniziale di crescita o la durata di quest'ultimo. In caso di attività di aaRS basso verso la ncAA, la sovraespressione dell'enzima endogeno o co-espressione di un diverso aaRS, che è più attivo verso la ncAA, possa essere condotte59.
L'aminoacido canonico Trp è dotato di tre caratteristiche notevoli: (i) la sua naturale abbondanza di proteine è basso; (ii) le sue proprietà biofisiche e chimici sono unici (ad es., è di solito l'origine dominante della fluorescenza intrinseca di proteine e peptidi) e (iii) contribuisce a una varietà di interazioni biochimiche e funzioni tra cui Π-stacking, H-legame interazioni catione-π. Tutte queste caratteristiche sono radicalmente cambiate su sostituzione di 4-ammino-Trp Trp → in dubbio GdFP. oltre, la progettazione di una classe di "oro" di avGFPs è un esempio notevole per proteine autofluorescent su misura di ingegneria. Con distinte proprietà spettrali, FPs può essere sintonizzato verso determinate finestre spettrale tramite incorporazione di mutagenesi e ncAA. In caso di GdFP, ciò avviene mediante un semplice scambio chimico H → NH2 nella cornice dell'indolo anello contenuta nella triade ECFP cromoforo. Figura 5 Mostra gli effetti di incorporazione di ncAA entro il cromoforo. L'introduzione del gruppo elettrone-donante provenienti da 4-ammino-indolo (intracellulare convertito in 4-ammino-Trp) consente una varietà di strutture mesomere che può spiegare uno stato eccitato stabilizzato. Spettroscopicamente, sua allargata spostamento di Stokes e l'emissione di fluorescenza rosso-spostato il risultato di queste proprietà distinte del sistema coniugato ampliato. Come riportato in precedenza, il trasferimento di carica intramolecolare avanzata entro il cromoforo GdFP è intrinsecamente sensibile al pH (Figura 4B) e accompagnata da un più grande cambiamento nel momento di dipolo tra terra0 S e S1 stato eccitato relativo a ECFP33. Come gruppi alternativi di elettrone-donante, analoghi di triptofano recanti un indolo sostituito con gruppi idrossi potrebbero essere utilizzati, come riportato in uno studio comparativo con il modello della proteina barstar41.
Gli spettri di assorbimento e fluorescenza di GdFP sono ampliati rispetto al ECFP ed EGFP (Figura 3 e D). Ampliamento omogeneo delle bande di assorbimento e fluorescenza è generalmente causata da modi vibrazionali nel cromoforo che, inoltre, dall'accoppiamento del cromoforo per ulteriori modi vibrazionali presenti nella proteina60. L'accoppiamento con l'ambiente locale della proteina è supportato da oneri localizzati il cromoforo. La disomogeneità strutturale della proteina porta a variazioni locali dello spettro vibronico, tale accoppiamento vibronico spettri del cromoforo e il resto della proteina sono supportati dalla delocalizzazione di carica e mesomere stati come indicato in Nella figura 5. Questo accoppiamento supporta anche il grande spostamento di Stokes e necessariamente riduce il rendimento quantico di fluorescenza. In confronto ad altri FPs rosso-spostato, GdFP esibisce anche stabilità proteica migliorata e una bassa tendenza alla aggregazione33,61,62. Non solo si differenzia nel colore dalle altre varianti della FP, ma espone anche una termostabilità sostanzialmente aumentato e migliorato cooperativa pieghevole33. L'intensità di fluorescenza è almeno il 90% conservato a riscaldamento a 60 ° C, mentre la fluorescenza ECFP è ridotto a circa il 30%. In proteine, aminoacidi aromatici spesso contribuiscono alle reti delle catene laterali interagenti, che comunemente hanno un effetto stabilizzante sulla struttura terziaria della proteina. avGFP porti tale lato catena rete, che consiste del cromoforo stesso, come bene come Phe-165, His-148 e Tyr-145. Queste catene laterali non sono solo abbastanza rigide in GdFP struttura33, ma la cosa importante, formano contatti idrofobici con il cromoforo. La caratteristica più importante romanzo identificata in GdFP è che il cromoforo amminato è più prossimale a Phe-165. Questa interazione è una caratteristica non osservata in altri noti avGFPs. Come due residui sono 3,2-4,5 Å apart, aminoacidi aromatici interazioni potrebbero essere anche presenti. Insieme con la stabilizzazione di risonanza amminazione-indotta del cromoforo, questi probabilmente stabilizzare questa rete idrofobica di aminoacidi in modo cooperativo. Un più efficace trasferimento di carica intramolecolare potrebbe essere supportato da queste interazioni nello stato eccitato rispetto allo stato di terra del cromoforo, e almeno in parte rappresenta il33,shift di Stokes 108 nm62 .
In progettazione razionale di proprietà fluoroforo, un aumento delle dimensioni del sistema π delocalizzato è preveduto per provocare una lunghezza d'onda di eccitazione rosso-spostato. Questa regola è obbedita dalla serie di aminoacidi in posizione 66 che porta ad neutri cromofori: Phe (λmax = 355 nm) < suo (λma x= 386 nm) < Tyr (λmax = 395 nm) < Trp (λmax = 436 nm)63. In natura, questa estensione del sistema coniugato di cromoforo di legami π è stato raggiunto da diverse strategie. Per DsRed da Discosoma striata, si estende dall'integrazione di un aminoacido ulteriore, spostando così λmax a 573 nm64. Il cromoforo della asFP595 (λmax = 595 nm) da Anemonia sulcata è stato esteso da un gruppo immino, allargando la sua π-sistema65. Poiché il cromoforo della GdFP e altri avFPs è della stessa dimensione, un principio diverso deve comportare una lunghezza d'onda di emissione nella gamma del DsRed espanso e asFP595 cromofori. Il profondo cambiamento di Stokes di 108 nm è attribuita alla struttura distinta del cromoforo GdFP, che rivela un nuovo principio fotofisiche nella progettazione di autofluorescent proteine. Calcoli preliminari (come segnalato nel 62) hanno dimostrato che il momento di dipolo del cromoforo allo stato eccitato di GdFP è sostanzialmente più grande nello stato fondamentale, in contrasto con i rispettivi valori di ECFP. Considerando che il momento di dipolo di GdFP aumenta da ~ 3 D (Debye) nello stato0 S a ~ 15 S1, il cambiamento per il cromoforo ECFP è piuttosto moderato (da ~ 4 D a ~ 6 D). Così, la fluorescenza dorata unica della GdFP è causata dal trasferimento di carica intramolecolare sostanziale entro il cromoforo, che aumenta la varietà di possibili strutture mesomere (Vedi Figura 5) che consentono di stabilizzazione di risonanza. Questo riduce il livello di energia da cui emissione si verifica. Come conseguenza il cambiamento profondo nel momento di dipolo all'eccitazione, la separazione di carica intramolecolare è il motivo principale per i cambiamenti nel potenziale elettrostatico dell'ambiente cromoforo. La matrice proteica circostante, a sua volta, regola ai cambiamenti nella distribuzione di carica dopo l'eccitazione del cromoforo. Il successivo rilassamento strutturale abbassa il livello di energia del cromoforo eccitato, che sposta lo spettro di fluorescenza al rosso grazie al suo carattere di trasferimento di carica. Per lo stesso motivo, in conseguenza del grande spostamento di Stokes e tariffe avanzate dei processi non radiativa, il rendimento quantico di fluorescenza di GdFP è ridotto rispetto al ECFP33.
Il rendimento quantico alto e piccolo spostamento di Stokes di ECFP ed EGFP sono solitamente attribuiti a un ambiente di proteina rigida del cromoforo, che riduce i gradi di libertà e, di conseguenza, conversione interna per favorire il rilassamento radiativo di stato eccitato 66. di conseguenza, il disegno molecolare di cromofori più rigidamente incorporati con accoppiamento ridotto a matrice della proteina rimanente potrebbe servire come una guida per produrre rosso-spostato più lontano di derivati GFP con rendimento quantico alta fluorescenza. Pertanto, per ulteriore ingegneria approcci per produrre proteine autofluorescent rosso-spostato, l'allargamento del sistema π-elettroni e una struttura rigida cromoforo con debole accoppiamento all'ambiente della proteina è altamente desiderabile. Tali modifiche potrebbero essere introdotti anche direttamente nella base di GFP cromofori o di collocamento di ncAAs desiderato nelle vicinanze cromoforo.
Disclosures
Gli autori dichiarano di non avere nessun concorrenti interessi finanziari.
Acknowledgments
Questo lavoro è stato supportato dalla German Research Foundation (Cluster di eccellenza "unificante concetti in catalisi) T.F. e N.B. e dal Ministero federale dell'educazione e della scienza (BMBF programma"HSP 2020", TU-WIMIplus progetto SynTUBio) di F.-J.S.
Materials
| Name | Company | Catalog Number | Comments |
| Chemicals | |||
| 4-aminoindole | Sigma-Aldrich | 525022 | |
| acetonitrile | VWR | HiPerSolv CHROMANORM ULTRA for LC-MS, 83642 | LC-MS grade required |
| agar-agar | Carl Roth | 5210 | |
| ammonium molybdate ((NH4)2MoO4) | Sigma-Aldrich | 277908 | |
| ammonium sulfate ((NH4)2SO4) | Sigma-Aldrich | A4418 | |
| ampicillin sodium salt | Carl Roth | K029 | |
| biotin | Sigma-Aldrich | B4501 | |
| bromophenol blue | Sigma-Aldrich | B0126 | |
| calcium chloride (CaCl2) | Sigma-Aldrich | C5670 | |
| colloidal silica | Sigma-Aldrich | Ludox HS-40, 420816 | |
| Coomassie Brillant Blue R 250 | Carl Roth | 3862 | |
| copper sulfate (CuSO4) | Carl Roth | CP86.1 | |
| D-glucose | Carl Roth | 6780 | |
| di-sodium hydrogen phosphate (Na2HPO4) | Carl Roth | X987 | |
| di-potassium hydrogen phosphate (K2HPO4) | Carl Roth | P749.1 | |
| 1,4-dithiothreitol (DTT) | Carl Roth | 6908 | |
| DNase I | Sigma-Aldrich | D5025 | |
| ethanol | Carl Roth | 9065.1 | |
| formic acid | VWR | HiPerSolv CHROMANORM for LC-MS, 84865 | LC-MS grade required |
| glycerol | Carl Roth | 3783 | |
| imidazole | Carl Roth | X998 | |
| indole | Sigma-Aldrich | I3408 | |
| iron(II) chloride (FeCl2) | Sigma-Aldrich | 380024 | |
| isopropanol | Carl Roth | AE73.1 | |
| isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | I6758 | |
| lysozyme | Sigma-Aldrich | L6876 | |
| magnesium chloride (MgCl2) | Carl Roth | KK36.1 | |
| magnesium sulfate (MgSO4) | Carl Roth | 8283.2 | |
| manganese chloride (MnCl2) | Sigma-Aldrich | 63535 | |
| β-mercaptoethanol | Carl Roth | 4227.3 | |
| potassium chloride (KCl) | Carl Roth | 6781.3 | |
| potassium dihydrogen phosphate (KH2PO4) | Sigma-Aldrich | P5655 | |
| RNase A | Carl Roth | 7156 | |
| sodium chloride (NaCl) | Carl Roth | P029 | |
| sodium dihydrogen phosphate (NaH2PO4) | Carl Roth | T879 | |
| sodium dodecyl sulphate (NaC12H25SO4) | Carl Roth | 0183 | |
| thiamine | Sigma-Aldrich | T4625 | |
| Tris(hydroxymethyl)-aminomethane (Tris) | Carl Roth | 5429 | |
| Tris hydrochloride (Tris-HCl) | Sigma-Aldrich | 857645 | |
| tryptone | Carl Roth | 8952 | |
| yeast extract | Carl Roth | 2363 | |
| zinc chloride (ZnCl2) | Sigma-Aldrich | 229997 | |
| Name | Company | Catalog Number | Comments |
| amino acids | |||
| L-alanine | Sigma-Aldrich | A7627 | |
| L-arginine | Sigma-Aldrich | A5006 | |
| L-asparagine | Sigma-Aldrich | A8381 | |
| L-aspartic acid | Sigma-Aldrich | A0884 | |
| L-cysteine | Sigma-Aldrich | C7352 | |
| L-glutamic acid | Sigma-Aldrich | G2128 | |
| L-glutamine | Sigma-Aldrich | G3126 | |
| L-glycine | Sigma-Aldrich | G7126 | |
| L-histidine | Sigma-Aldrich | H8000 | |
| L-isoleucine | Sigma-Aldrich | I2752 | |
| L-leucine | Sigma-Aldrich | L8000 | |
| L-lysine | Sigma-Aldrich | L5501 | |
| L-methionine | Sigma-Aldrich | M9625 | |
| L-proline | Sigma-Aldrich | P0380 | |
| L-phenylalanine | Sigma-Aldrich | P2126 | |
| L-serine | Sigma-Aldrich | S4500 | |
| L-threonine | Sigma-Aldrich | T8625 | |
| L-tryptophan | Sigma-Aldrich | T0254 | |
| L-tyrosine | Sigma-Aldrich | T3754 | |
| L-valine | Sigma-Aldrich | V0500 | |
| Name | Company | Catalog Number | Comments |
| Lab materials | |||
| 0.45 µm syringe filter with PVDF membrane | Carl Roth | CCY1.1 | |
| 1.5 mL microcentrifuge tubes | Eppendorf | 30120086 | |
| conical polystyrene (Falcon) tubes, 50 mL | Fisher Scientific | 14-432-22 | |
| Luer-Lock syringe 5 mL | Carl Roth | EP96.1 | |
| dialysis membrane, Molecular Weight Cut-Off (MWCO) 5,000 | Spectrum Medical Industries | Spectra/Por MWCO 5000 dialysis membrane, 133198 | |
| Immobilized Metal ion Affinity Chromatography (IMAC) column 1 mL, Ni-NTA | Macherey Nagel | Protino series, 745410.5 | |
| petri dishes (polystyrene, sterile) | Carl Roth | TA19 | |
| pQE-80L plasmid vector | Qiagen | no longer available | replaced by N-terminus pQE Vector set Cat No./ID: 32915 |
| protein extraction reagent BugBuster | EMB Millipore | 70921-4 | |
| round-bottom polystyrene tubes, 14 mL | Fisher Scientific | Corning Falcon, 14-959-1B | |
| Trp-auxotrophic E. coli strain | ATCC | ATCC 49980 | Bridges BA et al., Chem Biol Interact., 1972, 5(2):77-84; see main text for alternatives |
| Name | Company | Catalog Number | Comments |
| Mass Spectrometry equipment | |||
| mass spectrometer for LC-ESI-TOF-MS | Agilent | Agilent 6530 Accurate-Mass QTOF | coupled with Infinity LC system |
| mass spectrometry data analysis software | Agilent | MassHunter Qualitative Analysis software v. B.06.00 | |
| High-Performance Liquid Chromatography (HPLC) column for LC-ESI-TOF-MS | Sigma-Aldrich | Supelco Discovery BIO Wide Pore C5 HPLC column, 3 µm particle size, 10 cm x 2.1 mm | |
| HPLC autosampler vials 1.5 mL | Sigma-Aldrich | Supelco 854165 | with conical 0.1 mL glass inserts, screw caps and septa |
| Name | Company | Catalog Number | Comments |
| General equipment | |||
| benchtop centrifuge for 1.5 mL Eppendorf tubes | Eppendorf | 5427 R | |
| cooling centrifuge for 50 mL Falcon tubes | Eppendorf | 5810 R | |
| high pressure microfluidizer for bacterial cell disruption | Microfluidics | LM series with “Z” type chamber | |
| peristaltic pump for LC | GE Healthcare | P-1 | |
| Fast Protein Liquid Chromatography (FPLC) system | GE Healthcare | ÄKTA pure 25 L | |
| orbital shaker for bacterial cultivation | Infors HT | Minitron | |
| UV/Vis spectrophotometer | Biochrom | ULTROSPEC 2100 | |
| ultrasonic homogenizer for bacterial cell disruption | Omnilab | Bandelin SONOPULS HD 3200, 5650182 | with MS72 sonifier tip |
| Name | Company | Catalog Number | Comments |
| Fluorescence spectroscopy equipment | |||
| ps-pulsed laser 470 nm | Picoquant GmbH | PDL-470 | |
| time- and wavelength-correlated single photon counting (TWSPC) acquisition software | Picoquant GmbH | SymPhoTime 64 | |
| time- and wavelength-correlated single photon counting (TWSPC) detector | Picoquant GmbH | PML-16C | 16 spectral channels, to be selected by grating settings |
| single photon counting software | Picoquant GmbH | SPCM 9.75 | |
| global fitting software | Picoquant GmbH | SPC2Glo(R) | |
| fluorescence decay data analysis software | Picoquant GmbH | FluoFit program | |
| data analysis software | OriginLab Inc. | Origin 9.2 | |
| neutral density filter set | Schott | NG1 to NG11 | (400 - 650 nm, transmission 50 %, 20%, 10 %, 5 %) |
| 488 nm long-pass emission filter | AHF Analysentechnik | AHF-488 | |
| quartz cuvette | Thorlabs GmbH | CV10Q1400 | 1 cm pathlength |
References
- Shimomura, O., Johnson, F. H., Saiga, Y. Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea. J Cell Compar Physl. 59 (3), 223-239 (1962).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263 (5148), 802-805 (1994).
- Andresen, M., et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. P Natl Acad Sci USA. 102 (37), 13070-13074 (2005).
- Andresen, M., et al. Structural basis for reversible photoswitching in Dronpa. P Natl Acad Sci USA. 104 (32), 13005-13009 (2007).
- Brakemann, T., et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat Biotechnol. 29 (10), 942-947 (2011).
- Kremers, G. -J., Gilbert, S. G., Cranfill, P. J., Davidson, M. W., Piston, D. W. Fluorescent proteins at a glance. J Cell Sci. 124 (Pt 2), 157-160 (2011).
- Shimomura, O. Structure of the chromophore of aequorea 0. shimomura green fluorescent protein. FEBS Lett. 104 (2), 220-222 (1979).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 22 (12), 1567-1572 (2004).
- Shcherbo, D., et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 4 (9), 741-746 (2007).
- Shcherbakova, D. M., Subach, O. M., Verkhusha, V. V. Red fluorescent proteins: advanced imaging applications and future design. Angew Chem Int Edit. 51 (43), 10724-10738 (2012).
- Stepanenko, O. V., Verkhusha, V. V., Kuznetsova, I. M., Uversky, V. N., Turoverov, K. K. Fluorescent proteins as biomarkers and biosensors: throwing color lights on molecular and cellular processes. Curr Protein Pept Sc. 9 (4), 338-369 (2008).
- Wang, L., Xie, J., Deniz, A. A., Schultz, P. G. Unnatural amino acid mutagenesis of green fluorescent protein. J Org Chem. 68 (1), 174-176 (2003).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Sharma, N., Furter, R., Kast, P., Tirrell, D. A. Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 467 (1), 37-40 (2000).
- Liu, C. C., Schultz, P. G. Adding new chemistries to the genetic code. Annu Rev Biochem. 79, 413-444 (2010).
- Twine, S. M., Murphy, L., Phillips, R. S., Callis, P., Cash, M. T., Szabo, A. G. The Photophysical Properties of 6-Azaindole. J Phys Chem B. 107 (2), 637-645 (2003).
- Lepthien, S., Hoesl, M. G., Merkel, L., Budisa, N. Azatryptophans endow proteins with intrinsic blue fluorescence. P Natl Acad Sci USA. 105 (42), 16095-16100 (2008).
- Budisa, N., et al. Probing the role of tryptophans in Aequorea victoria green fluorescent proteins with an expanded genetic code. Biol Chem. 385 (2), 191-202 (2004).
- Ross, J. B., et al. Spectral enhancement of proteins: biological incorporation and fluorescence characterization of 5-hydroxytryptophan in bacteriophage lambda cI repressor. P Natl Acad Sci USA. 89 (24), 12023-12027 (1992).
- Soumillion, P., Jespers, L., Vervoort, J., Fastrez, J. Biosynthetic incorporation of 7-azatryptophan into the phage lambda lysozyme: Estimation of tryptophan accessibility, effect on enzymatic activity and protein stability. Protein Eng Des Sel. 8 (5), 451-456 (1995).
- Heim, R., Tsien, R. Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 6 (2), 178-182 (1996).
- Bridges, B. A., Mottershead, R. P., Rothwell, M. A., Green, M. H. L. Repair-deficient bacterial strains suitable for mutagenicity screening: tests with the fungicide captain. Chem Biol Interact. 5 (2), 77-84 (1972).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: The Heat Shock Method. J Vis Exp. , (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: Electroporation. J Vis Exp. , (2017).
- Grigorenko, B. L., Krylov, A. I., Nemukhin, A. V. Molecular modeling clarifies the mechanism of chromophore maturation in the green fluorescent protein. J Am Chem Soc. , (2017).
- JoVE Science Education Database. General Laboratory Techniques. Introduction to the Spectrophotometer. J Vis Exp. , (2017).
- Goedhart, J., et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 3, 751 (2012).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F. Culture medium for enterobacteria. J Bacteriol. 119 (3), 736-747 (1974).
- Hörnsten, E. G. On culturing Escherichia coli on a mineral salts medium during anaerobic conditions. Bioprocess Eng. 12 (3), 157-162 (1995).
- Davis, B. D. The Isolation of Biochemically Deficient Mutants of Bacteria by Means of Penicillin. P Natl Acad Sci USA. 35 (1), 1-10 (1949).
- Sambrook, J., Russell, D. W. Molecular Cloning: A Laboratory Manual. , Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY, USA. (2001).
- Wang, Y. -S., et al. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol Biosyst. 7 (3), 714-717 (2011).
- Bae, J. H., et al. Expansion of the genetic code enables design of a novel "gold" class of green fluorescent proteins. J Mol Biol. 328 (5), 1071-1081 (2003).
- JoVE Science Education Database. Dialysis: Diffusion Based Separation. J Vis Exp. , Cambridge, MA. (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Separating Protein with SDS-PAGE. J Vis Exp. , (2017).
- Petrásek, Z., et al. Excitation energy transfer from phycobiliprotein to chlorophyll d in intact cells of Acaryochloris marina studied by time- and wavelength-resolved fluorescence spectroscopy. Photoch Photobio Sci. 4 (12), 1016-1022 (2005).
- Kolber, Z. S., Barkley, M. D. Comparison of approaches to the instrumental response function in fluorescence decay measurements. Anal Biochem. 152 (1), 6-21 (1986).
- Pelet, S., Previte, M. J. R., Laiho, L. H., So, P. T. C. A fast global fitting algorithm for fluorescence lifetime imaging microscopy based on image segmentation. Biophys J. 87 (4), 2807-2817 (2004).
- Loefroth, J. E. Time-resolved emission spectra, decay-associated spectra, and species-associated spectra. J Phys Chem. 90 (6), 1160-1168 (1986).
- Hartman, M. C. T., Josephson, K., Lin, C. -W., Szostak, J. W. An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS One. 2 (10), e972 (2007).
- Budisa, N., et al. Global replacement of tryptophan with aminotryptophans generates non-invasive protein-based optical pH sensors. Angew Chem Int Edit. 41 (21), 4066-4069 (2002).
- Ma, Y., Biava, H., Contestabile, R., Budisa, N., di Salvo, M. L. Coupling bioorthogonal chemistries with artificial metabolism: intracellular biosynthesis of azidohomoalanine and its incorporation into recombinant proteins. Molecules. 19 (1), 1004-1022 (2014).
- Teramoto, H., Kojima, K. Incorporation of Methionine Analogues Into Bombyx mori Silk Fibroin for Click Modifications. Macromol Biosci. 15 (5), 719-727 (2015).
- Deal, R. B., Henikoff, J. G., Henikoff, S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 328 (5982), 1161-1164 (2010).
- Hinz, F. I., Dieterich, D. C., Tirrell, D. A., Schuman, E. M. Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci. 3 (1), 40-49 (2012).
- Dieterich, D. C., et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 13 (7), 897-905 (2010).
- Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A., Schuman, E. M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). P Natl Acad Sci USA. 103 (25), 9482-9487 (2006).
- Glenn, W. S., et al. Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT) Enables Time-Resolved Analysis of Protein Synthesis in Native Plant Tissue. Plant Physiol. 173 (3), 1543-1553 (2017).
- Zhou, L., et al. Incorporation of tryptophan analogues into the lantibiotic nisin. Amino Acids. 48 (5), 1309-1318 (2016).
- Acevedo-Rocha, C. G., Budisa, N. Xenomicrobiology: a roadmap for genetic code engineering. Microb Biotechnol. 9 (5), 666-676 (2016).
- Agostini, F., Völler, J. -S., Koksch, B., Acevedo-Rocha, C. G., Kubyshkin, V., Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew Chem Int Edit. 56 (33), 9680-9703 (2017).
- Bacher, J. M., Ellington, A. D. Selection and characterization of Escherichia coli variants capable of growth on an otherwise toxic tryptophan analogue. J Bacteriol. 183 (18), 5414-5425 (2001).
- Wong, J. T. Membership mutation of the genetic code: loss of fitness by tryptophan. Pc Natl Acad Sci USA. 80 (20), 6303-6306 (1983).
- Hoesl, M. G., et al. Chemical Evolution of a Bacterial Proteome. Angew Chem Int Edit. 54 (34), 10030-10034 (2015).
- Italia, J. S., et al. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat Chem Biol. 13 (4), 446-450 (2017).
- Völler, J. -S., Thi To, T. M., Biava, H., Koksch, B., Budisa, N. Global substitution of hemeproteins with noncanonical amino acids in Escherichia coli with intact cofactor maturation machinery. Enzyme Microb Tech. 106, 55-59 (2017).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Völler, J. -S., Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr Opin Biotech. 48, 1-7 (2017).
- Johnson, J. A., Lu, Y. Y., Van Deventer, J. A., Tirrell, D. A. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr Opin Chem Biol. 14 (6), 774-780 (2010).
- Somsen, O. J., van Grondelle, R., van Amerongen, H. Spectral broadening of interacting pigments: polarized absorption by photosynthetic proteins. Biophys J. 71 (4), 1934-1951 (1996).
- Kurschus, F. C., Pal, P. P., Bäumler, P., Jenne, D. E., Wiltschi, B., Budisa, N. Gold fluorescent annexin A5 as a novel apoptosis detection tool. Cytom Part A. 75 (7), 626-633 (2009).
- Lepthien, S., Wiltschi, B., Bolic, B., Budisa, N. In vivo engineering of proteins with nitrogen-containing tryptophan analogs. Appl Microbiol Biot. 73 (4), 740-754 (2006).
- Wachter, R. M., Elsliger, M. -A., Kallio, K., Hanson, G. T., Remington, S. J. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure. 6 (10), 1267-1277 (1998).
- Verkhusha, V. V., Lukyanov, K. A. The molecular properties and applications of Anthozoa fluorescent proteins and chromoproteins. Nat Biotechnol. 22 (3), 289-296 (2004).
- Martynov, V. I., Savitsky, A. P., Martynova, N. Y., Savitsky, P. A., Lukyanov, K. A., Lukyanov, S. A. Alternative cyclization in GFP-like proteins family. The formation and structure of the chromophore of a purple chromoprotein from Anemonia sulcata. J Biol Chem. 276 (24), 21012-21016 (2001).
- Piatkevich, K. D., Malashkevich, V. N., Morozova, K. S., Nemkovich, N. A., Almo, S. C., Verkhusha, V. V. Extended Stokes shift in fluorescent proteins: chromophore-protein interactions in a near-infrared TagRFP675 variant. Sci Rep. 3 (1), 1847 (2013).
