

Summary
Il s’agit d’une méthode pour générer des virus vaccinias recombinants « sans cicatrices » à l’aide de la sélection de la gamme d’hôtes et de l’identification visuelle des virus recombinants.
Abstract
Le virus vaccinia (VACV) a joué un rôle déterminant dans l’éradication du virus de la variole (VARV), l’agent causatif de la variole, de la nature. Depuis sa première utilisation comme vaccin, vaCV a été développé comme vecteur de vaccins thérapeutiques et de virus oncolytique. Ces applications tirent parti du génome facilement manipulé de VACV et de la large gamme d’hôtes comme plate-forme exceptionnelle pour générer des virus recombinés avec une variété d’applications thérapeutiques. Plusieurs méthodes ont été développées pour générer des VACV recombinés, y compris des méthodes de sélection de marqueurs et une sélection dominante transitoire. Ici, nous présentons un raffinement d’une méthode de sélection de gamme d’hôtes couplée à l’identification visuelle des virus recombinants. Notre méthode tire parti de la pression sélective générée par la protéine antivirale hôte kinase R (PKR) couplée à un gène de fusion fluorescent exprimant mCherry-tagged E3L, l’un des deux antagonistes VACV PKR. La cassette, y compris le gène d’intérêt et la fusion mCherry-E3L, est flanquée de séquences dérivées du génome du VACV. Entre le gène d’intérêt et mCherry-E3L est une région plus petite qui est identique aux premiers nucléotides de 150 pouces du bras 3', pour favoriser la recombinaison et la perte homologues du gène mCherry-E3L après sélection. Nous démontrons que cette méthode permet une génération efficace et transparente de vruvure dans une variété de types de cellules sans nécessiter la sélection de médicaments ou un dépistage approfondi des virus mutants.
Introduction
Le virus vaccinia (VACV) a joué un rôle déterminant dans la première éradication réussie d’un agent pathogène humain, le virus de la variole (VARV), de la nature. Depuis l’extermination du virus de la variole, les virus du vaxovirus, y compris le VVC, continuent d’être des virus thérapeutiques utiles pour la médecine humaine et animale. Par exemple, un vaccin contre le virus de la rage à base de VGRI a été très efficace pour prévenir la transmission de la rage sylvatique en Europe1 et aux États-Unis2. Plus récemment, les poxvirus recombinés exprimant une variété de molécules antitumorales (p. ex., anticorps à chaîne unique ou érythropoïétine humaine) ont connu un succès encourageant en tant qu’agents oncolytiques3,4,5. VACV est particulièrement attrayant en tant que vecteur parce qu’il est facilement favorable à la manipulation génétique, possède une large gamme d’hôtes, et il est stable dans une variété de conditions, permettant un transport facile et la viabilité du vaccin dans le domaine6,7. Bien que de multiples techniques aient été mises au point pour générer des VACV recombinés pour les expériences en laboratoire et la génération de vaccins, les stratégies actuelles visant à générer ces virus ont des limites notables.
Grâce à l’utilité du VVC, de multiples stratégies pour générer des virus recombinants ont été développées. La première stratégie utilise une recombinaison homologue pour introduire une cassette comprenant le transgène et un gène marqueur sélectionnable comme un gène de résistance aux antibiotiques. La cassette est flanquée de deux nucléotides (nt) ou de bras plus gros qui dirigent le gène vers un site spécifique du génome viral, qui est ensuite intégré de façon stable par des événements double crossover8,,9,10. Cette stratégie est rapide et efficace; cependant, il en résulte du matériel génétique supplémentaire sous la forme du gène marqueur qui peut produire des effets inattendus. En outre, il existe une limite supérieure pratique au nombre de transgènes qui peuvent être introduits limités par le nombre de marqueurs sélectionnables uniques disponibles. Les stratégies transitoires de sélection dominante (TDS) ont abordé cette question en facilitant la génération de virus recombinants « sans cicatrices» 11. Grâce à cette stratégie, un plasmide contenant un gène VACV mutant et un gène marqueur sélectionnable sont intégrés dans le génome viral, mais sans ADN VACV flanquant supplémentaire. Cette approche se traduit par l’intégration transitoire de l’ensemble du plasmide et la duplication du gène VACV à la suite de l’intégration par un seul événement de croisement. Cet intermédiaire est stable tant qu’il est maintenu sous pression de sélection, ce qui permet l’enrichissement de cette construction. Lorsque la sélection est supprimée, la duplication VACV permet un deuxième événement de croisement qui entraîne l’élimination du plasmide et la formation subséquente du type sauvage (wt) ou du virus recombinant dans un rapport approximatif de 50:50. Alors que le TDS génère des virus recombinés sans nécessiter l’introduction stable de l’ADN étranger, plusieurs clones de virus doivent être examinés pour la mutation prévue par l’analyse de séquençage, une étape potentiellement longue et coûteuse.
Ici, nous présentons une approche pour générer des poxvirus recombinés combinant les meilleurs aspects de chacune de ces approches, similaire à une approche qui a été décrite pour la réplication incompetent vaccinia modifié Ankara12,13,14. Cette stratégie combine la sélection visuelle et de la gamme d’hôtes pour générer rapidement des virus recombinants par des événements multisegments doubles, et par la suite éliminer le gène marqueur sélectionnable par recombinaison homologue. Cette approche permet la génération rapide de mutants négociés par une recombinaison homologue, avec la nature « sans cicatrice » des approches TDS, tout en n’exigeant pas une étape de dépistage ultérieure pour distinguer les virus sauvages de type et mutant. Notre méthode utilise également la sélection de la gamme hôte à la place de la sélection d’antibiotiques, éliminant le risque de changements phénotypiques induits chimiquement dans la lignée cellulaire. Pour cette approche, nous avons choisi d’utiliser la protéine antivirale hôte kinase R (PKR) comme agent sélectif pour générer recombinant VACV. PKR est exprimé comme un monomère inactif dans la plupart des types de cellules15. Sur la liaison arnorisation à double brin (dsRNA) dans les domaines de liaison N-terminal dsRNA, PKR dimerizes et est autophosphorylated16. Cette forme active de phosphorylates PKR l’alpha sous-unité du facteur d’initiation eucaryotique 2 (eIF2), inhibant finalement la livraison de l’initiateur méthionyl-tRNA au ribosome, empêchant ainsi la traduction intracellulaire et inhibant largement la réplication de nombreuses famillesvirales 17,18.
En réponse à l’activité antivirale large et puissante de PKR, de nombreux virus ont développé au moins une stratégie pour prévenir l’activation de la PKR. La plupart des poxvirus expriment deux antagonistes PKR, codés par les gènes E3L et K3L dans VACV, qui contrarier PKR par deux mécanismes distincts19. E3 empêche l’homodimerisation PKR en liant l’ARN à double brin20,21, tandis que K3 agit comme un inhibiteur pseudo-ubstrate en liant directement à PKR activé et inhibant ainsi l’interaction avec son substrat eIF222. Fait important, ces deux antagonistes de PKR n’inhibent pas nécessairement PKR de toutes les espèces. Par exemple, l’homolog K3 du virus de la variole du mouton a fortement inhibé le PKR des moutons, tandis que l’homolog de la variole du mouton E3 n’a pas montré d’inhibition considérable de PKR23,24. Dans cette étude, nous présentons une méthode d’utilisation de la pression sélective à médiation PKR combinée à la sélection de fluorescence pour générer un recombinant VACV supprimé pour E3L et K3L (VC-R4), qui ne peut pas reproduire dans les cellules compétentes PKR dérivées de diverses espèces. Ce virus recombinant fournit un excellent arrière-plan pour la génération rapide de virus recombinants exprimant des gènes sous contrôle du promoteur indigène E3L.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Générer le vecteur de recombinaison
- Amorce de conception pour générer la cassette de sélection. Concevez chaque amplicon individuel avec des séquences qui se chevauchent avec les amplicons voisins et le vecteur pour faciliter l’assemblage enzymatique isothermal des molécules d’ADN, également appelé assemblage Gibson, en utilisant l’un des outils de conception d’apprêt en ligne plusieurs.
REMARQUE : Ce protocole peut également être complété à l’aide de méthodes traditionnelles de clonage à base d’endonuclease de restriction. Dans ce cas, concevez des amorces avec les sites de restriction appropriés plutôt qu’avec des séquences qui se chevauchent. - À l’aide des amorces conçues à l’étape 1.1, PCR amplifie les éléments suivants dans l’ordre de 5' à 3' (figure 1) : 500 nucléotides de la région génomique VACV 5' de l’E3L (5' bras), de l’EGFP ou du gène d’intérêt, 150 nucléotides de la région génomique VACV immédiatement 3' de l’E3L (bras court 3'), un promoteur synthétique de poxvirus précoce/tardif25, le gène fusion mCherry-E3L, et 500 nucléotides de la région génomique VACV 3' de l’E3L dont le bras court 3' (long bras 3').
- Dans un tube PCR, ajouter les réactifs dans l’ordre suivant pour chaque amplicon : 17 ll d’eau libre DNase, 1,2 l de chaque amorce (concentration initiale de 10 m, concentration finale de 0,5 M), 5 L de tampon de réaction PCR 5x, ADN de modèle (10 ng pour les amplicons amplifiés à partir de plasmides : EGFP et E/L promoteur-mCherry-E3L cassette; 100 ng pour les amplicons amplifiés à partir de l’ADN génomique viral : 5' et 3' bras), et 0.5 'L de polymerase. Ajuster le volume d’eau ajouté pour un volume de réaction final de 50 ll.
REMARQUE : La concentration de l’ADN de modèle devrait être déterminée empiriquement, mais nous commençons généralement par 10 ng/réaction. - Placer le tube(s) dans un thermocycler, et faire fondre l’ADN à 98 oC pour 30 s, puis utiliser 25 tours d’un protocole PCR en trois étapes: 98 oC pour 5 s, 55 oC pour 10 s, et 72 oC pour 1 min.
REMARQUE : Déterminez la température de fonte en fonction du Tm suggéré par le fabricant pour chaque ensemble d’apprêt. Déterminez le temps de prolongation approprié en fonction de la durée de chaque amplicon (1 minute/kb).
- Dans un tube PCR, ajouter les réactifs dans l’ordre suivant pour chaque amplicon : 17 ll d’eau libre DNase, 1,2 l de chaque amorce (concentration initiale de 10 m, concentration finale de 0,5 M), 5 L de tampon de réaction PCR 5x, ADN de modèle (10 ng pour les amplicons amplifiés à partir de plasmides : EGFP et E/L promoteur-mCherry-E3L cassette; 100 ng pour les amplicons amplifiés à partir de l’ADN génomique viral : 5' et 3' bras), et 0.5 'L de polymerase. Ajuster le volume d’eau ajouté pour un volume de réaction final de 50 ll.
- Visualisez les produits d’amplification sur un gel agarose de 1 %. Ajouter 10 l de chaque produit d’ADN et 2 l de tampon de chargement à chaque puits, et fonctionner à 8 V/cm pendant 1 h.
- Gel purifiez chaque amplicon à l’aide d’un kit d’extraction de gel d’ADN et du protocole du fabricant. Elute les amplicons de la colonne en ajoutant 50 L d’eau libre DNase et immédiatement centrifugage.
- Linéariser le vecteur de clonage pUC19 à l’aide de la digestion de l’endonuclease EcoRI. À un tube, ajouter 1 g de pUC19, de l’eau à un volume de 17 ll, 2 L de tampon de réaction et 1 l (20 unités) d’EcoRI. Incuber à 37 oC pour 1 h.
- Visualisez les produits d’amplification sur un gel d’agarose de 1 % exécuté à 8 V/cm pendant 1 h. Excisez la bande du gel, et purifiez le produit à l’aide du kit d’extraction de gel d’ADN tel que décrit à l’étape 1.4.
- Ligater tous les amplicons individuels, gel purifié et le vecteur linéaire à l’aide d’un kit de mélange maître.
- Sur un tube PCR, ajouter 0,2 hol de pUC19 linéaire et chaque amplicon (5' bras, EGFP, bras court 3', cassette E/L promoteur-mCherry-E3L, 3'arm). Ajouter l’eau sans DNase à un volume final de 10 l, puis ajouter 10 l de mélange de maître d’assemblage d’ADN. Incuber des échantillons à 50 oC pour 1 h.
- Transformez E. coli chimiquement compétent avec 2 l du produit assemblé de l’étape 1.6 comme décrit précédemment26,27. Plaque 100 L des cellules transformées sur des plaques d’agarose LB contenant 100 g/mL d’ampicilline. Incuber les plaques pendant la nuit à 37 oC.
- Choisissez des colonies bien isolées et transférez des colonies individuelles dans des tubes contenant du bouillon Luria avec une ampicilline de 100 g/mL. Incuber les tubes pendant la nuit à 37 oC tout en secouant à 225 tr/min.
- Isolez les plasmides de la culture du jour au lendemain à l’aide d’un kit de miniprep plasmide. Vérifiez la concentration et la pureté de l’ADN à l’aide d’un spectrophotomètre. Un ratio A260/A280 compris entre 1,8 et 2,0 est acceptable.
- Soumettez les plasmides pour le séquençage Sanger pour déterminer si le produit de clonage désiré est correct. Conservez l’ADN à -20 oC.
2. Générer le virus recombinant
- Infecter un monolayer confluent de cellules appropriées avec le virus à recombiner à une multiplicité d’infection de 1,0 (MOI 1,0) dans une plaque de 6 puits. Incuber les cellules infectées à 37 oC et 5% de CO2 pour 1 h. Ensuite, aspirer le milieu et le remplacer par du DMEM frais. Incuber les cellules infectées à 37 oC et 5% de CO2.
REMARQUE : Pour les virus compétents de réplication tels qu’un virus vaccinia qui n’a pas de K3L22, une lignée cellulaire telle que la lignée européenne de cellules rénales de lapin RK13 (ATCC #CCL-37) ou BSC-40 est appropriée. Cependant, pour les virus déficients de réplication, tels que le virus décrit dans cet article manquant à la fois PKR antagonistes E3L et K3L, une ligne cellulaire complémentaire exprimant ces deux gènes dans les cellules trans ou PKR knock-down ou knock-out sont nécessaires. - Transfect les cellules infectées avec 500 ng du vecteur généré et validé à l’étape 1.10 à l’aide d’un réactif transfection disponible dans le commerce suivant le protocole du fabricant. Incuber les cellules à 37 oC et 5% de CO2 pour 48 h.
REMARQUE : Si l’utilisation d’un virus vacciniane dépourvu de la protéine de fusion E3L et K3L, la pression sélective à médiation PKR stimulera la sélection des virus recombinés et maintiendra l’expression de la protéine de fusion mCherry-E3L dans ces cellules. Si désiré, il devrait également être possible de PCR amplifier seulement l’insert à utiliser pour la transfection au lieu de l’ensemble du plasmide. - 48 heures après l’infection, récoltez le monolayer infecté. Dans certains cas, les cellules peuvent être récoltées par tuyauterie, mais si elles sont encore étroitement adhérées, récoltez-les avec un grattoir cellulaire. Congeler les cellules trois fois, puis sonicate les lysates pour 15 s à 50% d’amplitude. Conservez ce lysate à -80 oC jusqu’à ce qu’il soit prêt à l’emploi.
- Siluer en série 10 fois le lysate récolté à l’étape 2.3 de 10-1 à 10-6 en ajoutant 120 L du lysate à 1080 6 L de DMEM (10-1), puis en ajoutant 120 L de cette dilution à 1080 L de DMEM (10-2), et en répétant ce processus quatre fois de plus. Ajouter 1 ml de chaque dilution à un puits individuel et confluent d’une lignée cellulaire compétente de PKR, dans ce cas RK13 cellules.
- Incuber les cellules infectées à 37 oC et 5% de CO2 pour 1 h. Ensuite, aspirez le milieu et remplacez-le par DMEM frais Incuber les cellules infectées à 37 oC et 5% de CO2.
- 24 à 48 heures après l’infection, identifier les virus recombinants par microscopie de fluorescence. Les plaques des virus recombinants expriment la fluorescence rouge due à l’intégration du gène de fusion mCherry-E3L(figure 2). Si un virus dépourvu d’inhibiteurs de la PKR a été utilisé au départ, toutes les plaques contiendront le virus recombinant.
- Plaque purifier les virus recombinants trois fois sur les cellules RK13. Après la dernière ronde de purification de la plaque, toutes les plaques doivent exprimer la fluorescence rouge.
- Infectez une plaque confluente de cellules RK13 6-puits exprimant les inhibiteurs de VACV PKR E3L et K3L (cellules RK13-E3L-K3L28) avec le virus de fluorage rouge tri-purifié de l’étape 2.6. Visez environ 50-100 plaques par puits.
REMARQUE : Ces cellules fournissent les antagonistes DE VACV PKR dans les trans et allègent la pression sélective PKR-négociée pour maintenir le gène de fusion mCherry-E3L, favorisant ainsi la génération « sans cicatrice » du virus recombinant. - Identifier les virus effondrés par microscopie à fluorescence à l’aide d’un microscope EVOS2, et d’un cube de filtre GFP (Excitation: 470/22, Émission: 525/50) et un cube de filtre de DP (Excitation: 531/40, Émission: 593/40).
REMARQUE : La fréquence à laquelle le gène de fusion mCherry-E3L est perdu est d’environ 2,5 %(tableau 2). Si l’EGFP n’est pas incluse comme gène marqueur, les plaques des virus mutants qui ont perdu le gène de fusion mCherry-E3L seront incolores. - La plaque purifie trois fois les plaques vertes seulement (VC-R4) ou incolores (E3L) sur les cellules RK13-E3L-K3L. Assurez-vous qu’aucune plaque ne fluore rouge.
- Confirmer la perte de mCherry-E3L et la présence de la mutation attendue par le séquençage PCR et Sanger.
REMARQUE : Si le gène ou la mutation d’intérêt n’a pas d’activité inhibitrice de la PKR, les virus recombinés doivent être cultivés sur des cellules RK13-E3L-K3L ou sur une lignée cellulaire équivalente inhibée par le PKR ou en PKR(figure 3).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Nous avons utilisé la procédure diagrammed dans la figure 1 pour générer un VACV manquant à la fois PKR antagonistes E3L et K3L, en remplaçant E3L par EGFP dans un virus déjà supprimé pour K3L (vP872). La figure 2 montre des plaques fluorescentes rouges dans les cellules RK13 compétentes de PKR indicatifs de l’expression virale de mCherry-E3L, ainsi que l’EGFP exprimées dans les cellules RK13-E3L-K3L confirmant la perte de l’E3L et l’effondrement du marqueur de sélection mCherry-E3L. La figure 3 confirme que ce virus recombinant, VC-R4, dépourvu des deux antagonistes de PKR ne peut pas se répliquer dans les cellules RK13 compétentes de PKR, tandis que le virus parent, vP872 exprimant L’E3L, est de réplication compétente. Pour confirmer que cette incapacité à se répliquer dans les cellules RK13 n’était due qu’à la perte d’E3L, nous avons remplacé l’EGFP en VC-R4 par E3L, pour générer un virus de retour en utilisant le même protocole de sélection. La figure 3 valide également que ce virus de retour se reproduit aussi efficacement que le vP872 dans les cellules RK13. Fait intéressant, des plaques incolores compatibles à l’effondrement du marqueur de sélection mCherry-E3L ont été identifiées avant la sélection dans les cellules RK13-E3-K3 qui sont généralement nécessaires pour sélectionner des recombinaisons « sans cicatrice », probablement en raison de l’identité de séquence prolongée entre la cassette de recombinaison mCherry-E3L et le gène E3L étant inséré dans VC-R4. Par conséquent, pour déterminer l’efficacité de la recombinaison et le taux d’effondrement que nous avons choisi de produire des virus exprimant le poxvirus PKR antagoniste K3L pour éviter le problème de l’effondrement précoce23. La figure 4 indique l’apparition de plaques incolores (pointes de flèche) après l’infection des cellules RK13-E3L-K3L. Le tableau 1 montre les résultats de trois expériences indépendantes, où en moyenne 12,6 % des virions progénitures avaient subi une recombinaison avec le plasmide transfected, semblable aux fréquences précédemment signalées29,30,31. Le tableau 2 détaille la fréquence des plaques incolores par rapport aux plaques totales dans les cellules RK13-E3L-K3L, ce qui démontre le taux d’effondrement et la perte du marqueur de sélection mCherry-E3L s’est produit à une fréquence d’environ 1,8 %.
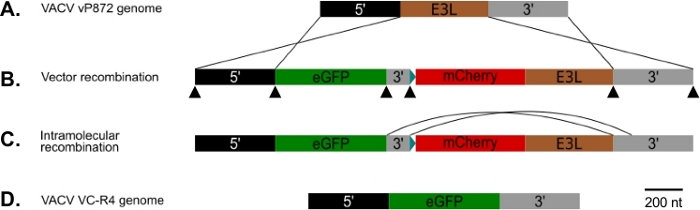
Figure 1 : Diagramme de p837-GOI-mCherry-E3L ainsi que la stratégie de recombinaison visuelle et de portée d’accueil. (A) 5' bras (noir) et 3' bras (gris) flanquent le locus E3L (brun) en VACV. (B) En p837-GOI-mCherry-E3L, ces bras flanquent une cassette contenant le gène d’intérêt (GOI), en l’occurrence EGFP, (vert) séparé d’un gène fusion mCherry-E3L (rouge) sous contrôle du promoteur synthétique du poxvirus précoce/tardif25 bleu) par un court bras de 3 po (gris). Ces bras externes conduisent la recombinaison homologue entre VACV et le p837-GOI-mCherry-E3L. Les pointes de flèche noires indiquent les emplacements des amorces qui se chevauchent utilisées pour générer ce plasmide par le clonage Gibson. (C) Lorsque la pression sélective PKR est enlevée, les virus qui ont subi une recombinaison intramoléculaire entre les bras courts et longs de 3 po peuvent être sélectionnés. (D) Résultant en un virus (VC-R4) contenant seulement le gène d’intérêt dans le locus E3L. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 2 : Micrographes fluorescents de (en haut) une plaque de virus recombinante 24 heures après recombinaison avec p837-GOI-mCherry-E3L exprimant à la fois mCherry (gauche) et EGFP (à droite) dans les cellules RK13. (En bas) Micrographe d’une plaque de virus recombinante 48 heures après que la pression sélective à médiation PKR a été enlevée dans les cellules RK13MD, exprimant l’EGFP (à droite) mais pas mCherry (à gauche). La barre d’échelle indique 650 m pour tous les panneaux. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
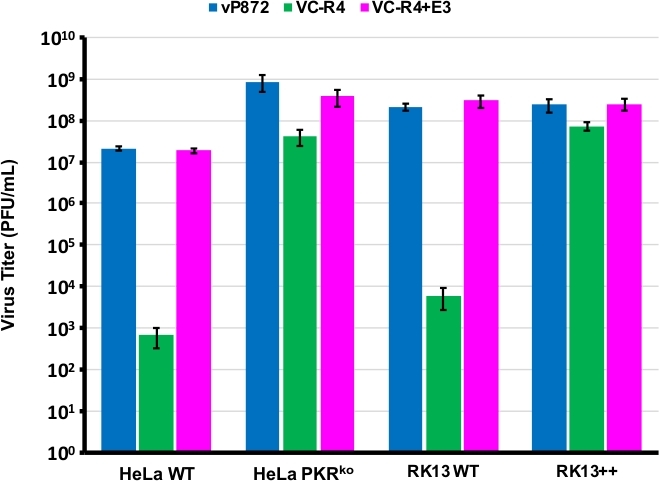
Figure 3 : VC-R4 ne peut pas se répliquer dans les cellules compétentes de PKR. Les lignées cellulaires indiquées ont été infectées par le vP872 (bleu), le VC-R4 (vert) ou le VC-R4-E3L (magenta) à MOI 0,1. 48 heures après l’infection, les cellules infectées ont été récoltées et titerées par dilution en série sur les cellules RK13-E3L-K3L. Les titres sont signalés dans PFU/mL, les barres d’erreurs représentent l’écart standard de trois expériences de réplique. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 4 : Perte d’expression mCherry-E3L dans les cellules RK13-E3L-K3L. Superposition de micrographes fluorescents et de contraste de phase des cellules VC-R4-K3L-mCherry-E3L infectées. Trois plaques n’expriment plus mCherry (cercles) en raison de l’effondrement de la cassette de sélection donnant VC-R4-K3L. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
| Expérience 1 | Expérience 2 | Expérience 3 | |
| Plaques rouges (RK13) | 30 | 11 | 18 |
| Plaques totales (RK13-E3L-K3L) | 225 | 64 | 249 |
| Taux de recombinaison | 13.30% | 17.20% | 7.20% |
Tableau 1 : Fréquence de recombinaison de VACV avec le plasmide p837-K3L-mCherry-E3L.
| Expérience 1 | Expérience 2 | Expérience 3 | |
| Plaques totales (RK13-E3L-K3L) | 115 | 44 | 210 |
| Plaques incolores (RK13-E3L-K3L) | 3 | 1 | 1 |
| Taux de recombinaison | 2.60% | 2.30% | 0.50% |
Tableau 2 : Fréquence de la perte de mCherry-E3L provenant des cellules VC-R4-K3L-mCherry-E3L dans les cellules RK13-E3-K3.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Ici, nous présentons une variation d’une stratégie transitoire de sélection des marqueurs 32 pour générer des virus vaccinia recombinants sans retenir l’ADN étranger dans le virus recombinant final. Notre stratégie utilise une pression sélective négociée par la protéine antivirale hôte PKR plutôt que d’autres formes de pression sélective comme les antibiotiques. L’utilisation de gènes antiviraux hôtes élimine la possibilité de changements phénotypiques induits chimiquement dans les cellules, ou un risque accru de mutation due à des médicaments de sélection. En outre, contrairement à la sélection des médicaments, il n’y a pas de phase de décalage pour notre approche, parce que PKR est exprimé de façon constitutive dans toutes les cellules. La sélection visuelle secondaire basée sur l’expression mCherry améliore également la spécificité de cette méthode en veillant à ce que seules les plaques exprimant le transgène soient cueillies au cours de la première phase, et soit tout aussi efficaces qu’un marqueur sélectif négatif tout en sélectionnant les virus recombinants matures qui ont perdu le gène mCherry-E3L.
Les étapes les plus cruciales de cette stratégie de recombinaison sont la génération du vecteur de recombinaison approprié, et la purification appropriée de la plaque pour s’assurer que le virus sélectionné est clonal. Dans cet article, nous suggérons "assemblage Gibson" pour générer le vecteur de recombinaison. Cette stratégie est extrêmement efficace et permet l’assemblage de tous les fragments comprenant le vecteur de recombinaison en une seule journée. Cependant, parce que le bras court de 3' et le bras long 3' partagent des séquences identiques, ces fragments ont le potentiel d’être réunis pendant la réaction de clonage, et certains vecteurs peuvent ne pas contenir la cassette mCherry-E3L. D’après notre expérience, c’est rare, mais il est nécessaire de confirmer la structure du vecteur après le clonage. Nous avons également généré des vecteurs de recombinaison pour cette stratégie en utilisant des méthodes traditionnelles d’endonuclease et de ligase. Cette stratégie évite le problème décrit ci-dessus, mais peut être plus exigeant en main-d’œuvre. La purification de la plaque est généralement simple et dépend principalement de l’utilisation de cellules permissives appropriées pour la recombinaison initiale, des cellules PKR-compétentes pour la purification initiale de la plaque pour s’assurer que seuls les virus recombinants peuvent se répliquer, puis les cellules permissives à nouveau pour faciliter la recombinaison intramoléculaire et la perte du marqueur sélectionnable. Une attention particulière aux lignées cellulaires est donc essentielle pour l’application réussie et efficace de cette stratégie.
Dans cette étude, nous démontrons l’utilisation de cette méthode pour générer un recombinant VACV supprimé pour les antagonistes PKR E3L et K3L et exprimant EGFP sous le contrôle du promoteur E3L. À l’avenir, ce virus servira de fond efficace pour les futurs virus recombinants, car il est incapable de se répliquer dans les cellules compétentes PKR. Par conséquent, il y aura une forte pression sélective PKR-négociée pour conduire la cassette de recombinaison mCherry-E3L dans les virions progénitures tout en empêchant essentiellement la réplication du virus non recombinant. En outre, la perte de l’EGFP par l’absorption de la cassette de recombinaison est un marqueur de sélection secondaire utile pour s’assurer que les plaques cueillies ne sont pas co-infectées par un virus non recombinant. Nous avons observé des taux de recombinaison compatibles avec les taux précédemment rapportés pour VACV, mais les marqueurs fluorescents visuels augmentent l’efficacité de générer des virus recombinés en s’assurant que l’augmentation de la probabilité que les virus recombinants appropriés sont sélectionnés. Notre observation des plaques incolores après deux tours de sélection sur les cellules PKR-compétentes, probablement en raison de la durée accrue de la séquence identique entre E3L et le gène marqueur mCherry-E3L, suggère que le taux de perte mCherry-E3L peut être « réglé » en augmentant ou en diminuant la longueur du bras court de 3. La principale limitation de cette technique est l’utilisation de PKR comme pression sélective pour les recombinants. L’utilisation la plus efficace de cette stratégie de recombinaison est de générer ces virus dans un contexte dépourvu d’antagonistes PKR. Cependant, le marqueur de sélection colorimétrique permet à cette stratégie de recombinaison d’être utilisée même sans la sélection médiée par PKR, simplement par plaque purifiant mCherry-exprimant plaques. Bien que l’absence de pression sélective à médiation PKR réduira l’efficacité de la première étape de dépistage, le pourcentage de plaques d’expression mCherry est encore assez élevé que la sélection basée sur les couleurs est viable. Ainsi, cette méthode peut être utilisée pour insérer presque n’importe quel gène dans le génome du virus du poxvirus.
Comme le démontre l’insertion de l’EGFP, avec cette approche, n’importe quel gène peut être rapidement inséré dans le locus E3L sous contrôle du promoteur indigène, à condition que les cellules nulles PKR ou les lignes cellulaires de compliment sont utilisées pour des expériences en aval si le transgène n’est pas un antagoniste de PKR. Cette stratégie, combinée au virus VC-R4 que nous rapportons ici, ajoute une méthode nouvelle et puissante pour générer rapidement et de façon fiable des virus vaccinias recombinants à l’aide de pression sélective à médiation d’hôte et d’identification visuelle des recombinaisons au début du processus.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs ne déclarent aucun intérêt financier concurrent.
Acknowledgments
Ce projet a été financé par les National Institutes of Health (AI1114851) à SR.
Materials
| Name | Company | Catalog Number | Comments |
| 2X-Q5 Master Mix | NEB | M0492L | High-fidelity polymerase used in PCR |
| Ampicillin | ThermoFisher Scientific | 11593027 | Bacterial selective agent |
| Disposable Cell Scrapers | ThermoFisher Scientific | 08-100-242 | Cell scraper to harvest infected cells |
| EVOS FL Auto 2 Cell imaging system | ThermoFisher Scientific | AMAFD2000 | Fluorescent microscope |
| EVOS Light Cube, GFP | ThermoFisher | AMEP4651 | GFP Cube |
| EVOS Light Cube, RFP | ThermoFisher | AMEP4652 | RFP Cube |
| GenJet | SignaGen Laboratories | SL100489 | Transfection reagent |
| Luria Bertani (LB) Broth | Gibco | 10855021 | Bacterial growth medium |
| Monarch DNA gel extraction kit | NEB | T1020L | Gel purification kit used to purify amplicons and linearized vectors |
| Monarch Plasmid Miniprep kit | NEB | T1010L | Miniprep kit ussed to purify plasmids |
| NanoDrop One | ThermoFisher Scientific | ND-ONE-W | Spectrophotometer used to measure RNA and DNA concentration |
| NEBuilder Master Mix | NEB | E2621L | Isothermal enzymatic assembly kit used to generate the recombination vector |
| Q500 Sonicator | Qsonica | Q500-110 | Sonicator for virus lysates |
| RK13 cells | ATCC | CCL-37 | Rabbit kidney cells |
| VWR Multiwell Cell Culture plates | VWR | 10062-892 | Cell culture plates |
References
- Brochier, B., et al. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature. 354 (6354), 520-522 (1991).
- Pastoret, P. P., Brochier, B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; a link between Jenner and Pasteur. Epidemiology and Infection. 116 (3), 235-240 (1996).
- Chan, W. M., McFadden, G.
- Nguyen, D. H., et al. Vaccinia virus-mediated expression of human erythropoietin in tumors enhances virotherapy and alleviates cancer-related anemia in mice. Molecular Therapy. 21 (11), 2054-2062 (2013).
- Frentzen, A., et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proceedings of the National Academy of Sciences of the United States of America. 106 (31), 12915-12920 (2009).
- Pastoret, P. P., Vanderplasschen, A.
- COLLIER, L. H. The development of a stable smallpox vaccine. The Journal of Hygiene. 53 (1), 76-101 (1955).
- Weir, J. P., Bajszár, G., Moss, B. Mapping of the vaccinia virus thymidine kinase gene by marker rescue and by cell-free translation of selected mRNA. Proceedings of the National Academy of Sciences of the United States of America. 79 (4), 1210-1214 (1982).
- Mackett, M., Smith, G. L., Moss, B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proceedings of the National Academy of Sciences of the United States of America. 79 (23), 7415-7419 (1982).
- Nakano, E., Panicali, D., Paoletti, E. Molecular genetics of vaccinia virus: demonstration of marker rescue. Proceedings of the National Academy of Sciences of the United States of America. 79 (5), 1593-1596 (1982).
- Falkner, F. G., Moss, B. Transient dominant selection of recombinant vaccinia viruses. Journal of Virology. 64 (6), 3108-3111 (1990).
- Staib, C., Drexler, I., Ohlmann, M., Wintersperger, S., Erfle, V., Sutter, G. Transient Host Range Selection for Genetic Engineering of Modified Vaccinia Virus Ankara. BioTechniques. 28 (6), 1137-1148 (2000).
- Staib, C., Drexler, I., Sutter, G. Construction and Isolation of Recombinant MVA. Vaccinia Virus and Poxvirology. , 77-99 (2004).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Pfaller, C. K., Li, Z., George, C. X., Samuel, C. E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Current Opinion in Immunology. 23 (5), 573-582 (2011).
- Bevilacqua, P. C., George, C. X., Samuel, C. E., Cech, T. R. Binding of the protein kinase PKR to RNAs with secondary structure defects: Role of the tandem A - G mismatch and noncontigous helixes. Biochemistry. 37 (18), 6303-6316 (1998).
- Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., Hinnebusch, A. G. Tight Binding of the Phosphorylated Subunit of Initiation Factor 2 (eIF2) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Molecular and Cellular Biology. 21 (15), 5018-5030 (2001).
- Rothenburg, S., Georgiadis, M. M., Wek, R. C. Evolution of eIF2α kinases: Adapting translational control to diverse stresses. Evolution of the Protein Synthesis Machinery and Its Regulation. , 235-260 (2016).
- Bratke, K. A., McLysaght, A., Rothenburg, S. A survey of host range genes in poxvirus genomes. Infection, Genetics and Evolution. 14, 406-425 (2013).
- Chang, H. W., Watson, J. C., Jacobs, B. L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proceedings of the National Academy of Sciences. 89 (11), 4825-4829 (1992).
- Romano, P. R., et al. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Molecular and Cellular Biology. 18 (12), 7304-7316 (1998).
- Beattie, E., Tartaglia, J., Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 183 (1), 419-422 (1991).
- Park, C., Peng, C., Brennan, G., Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Annals of the New York Academy of Sciences. 1438 (1), 18-29 (2019).
- Rothenburg, S., Brennan, G. Species-Specific Host-Virus Interactions: Implications for Viral Host Range and Virulence. Trends in Microbiology. , (2019).
- Chakrabarti, S., Sisler, J. R., Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 23 (6), 1094-1097 (1997).
- Chung, C. T., Niemela, S. L., Miller, R. H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution (recombinant DNA). Biochemistry. 86, 2172-2175 (1989).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Methods in Enzymology. 218, 621-627 (1993).
- Rahman, M. M., Liu, J., Chan, W. M., Rothenburg, S., McFadden, G. Myxoma Virus Protein M029 Is a Dual Function Immunomodulator that Inhibits PKR and Also Conscripts RHA/DHX9 to Promote Expanded Host Tropism and Viral Replication. PLOS Pathogens. 9 (7), 1003465 (2013).
- Evans, D. H., Stuart, D., McFadden, G. High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus-infected mammalian cells. Journal of Virology. 62 (2), 367-375 (1988).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. Journal of Virology. 61 (6), 1788-1795 (1987).
- Spyropoulos, D. D., Roberts, B. E., Panicali, D. L., Cohen, L. K. Delineation of the viral products of recombination in vaccinia virus-infected cells. Journal of Virology. 62 (3), 1046-1054 (1988).
- Liu, L., et al. Transient dominant host-range selection using Chinese hamster ovary cells to generate marker-free recombinant viral vectors from vaccinia virus. BioTechniques. 62 (4), 183-187 (2017).
