

Summary
これは、組み換えウイルスの宿主範囲選択および視覚的同定を用いて「瘢痕のない」組換えワクシニアウイルスを生成する方法である。
Abstract
ワクシニアウイルス(VACV)は天然痘の原因物質であるバリオラウイルス(VARV)を自然から根絶するのに役立った。ワクチンとして初めて使用されて以来、ワクチンのベクターとして、また、癌性ウイルスとして開発されてきました。これらのアプリケーションは、VACVの容易に操作されたゲノムおよび広い宿主範囲を優れたプラットフォームとして利用し、様々な治療用途を有する組換えウイルスを生成する。マーカー選択法や過渡性優先選択を含む組換えVACVを生成するためにいくつかの方法が開発されています。ここでは、組換えウイルスの視覚的同定と相まって、宿主範囲選択方法の改良を提示する。我々の方法は、宿主抗ウイルスタンパク質キナーゼR(PKR)によって生成される選択的圧力を、2つのVACV PKRアンタゴニストの1つであるmCherryタグ付きE3Lを発現する蛍光融合遺伝子と相まって利用する。カセットには、対象遺伝子およびmCherry-E3L融合を含む、VACVゲノムに由来する配列によって横たわっている。対象遺伝子とmCherry-E3Lとの間には、3'アームの最初の〜150ヌクレオチドと同一である小さい領域であり、選択後のmCherry-E3L遺伝子の相同組換えおよび喪失を促進する。我々は、この方法が、薬物選択や変異型ウイルスの広範なスクリーニングを必要とせずに、様々な細胞型における効率的でシームレスなrVACVの生成を可能にすることを実証する。
Introduction
ワクシニアウイルス(VACV)は、自然からヒト病原体、バリオラウイルス(VARV)の最初の成功した根絶に役立ちました。バリオラウイルスの駆除以来、VACVを含むポックスウイルスは、ヒトおよび動物医学の両方に有用な治療ウイルスであり続けています。例えば、VACVベースの狂犬病ウイルスワクチンは、欧州1及び米国2におけるシルベージ狂犬病の感染を予防するのに非常に有効であった。さらに最近では、様々な抗腫瘍分子(例えば、単鎖抗体またはヒトエリスロポエチン)を発現する組換えポックスウイルスは、腫瘍分解剤33、4、54,5として励ましの成功を見ている。VACVは、遺伝子操作に容易に適性であるためベクターとして特に魅力的であり、広い宿主範囲を有し、かつ様々な条件下で安定であり、フィールド66,77における容易な輸送およびワクチンの生存率を可能にする。実験室実験やワクチン生成のための組換えVACVを生成する複数の技術が開発されているが、これらのウイルスを生成するための現在の戦略には顕著な限界がある。
VACVの有用性により、組換えウイルスを生成する複数の戦略が開発されている。第1の戦略は、抗生物質耐性遺伝子などのトランスジーンおよび選択可能マーカー遺伝子を含むカセットを導入するために相同組換えを採用する。カセットは、2〜500ヌクレオチド(nt)以上の腕によって横にされ、遺伝子をウイルスゲノムの特定の部位に導き、その後、二重交差事象88、9、109,10によって安定的に統合される。この戦略は迅速かつ効率的です。しかし、それは予期しない効果を生み出す可能性のあるマーカー遺伝子の形で余分な遺伝物質をもたらす。さらに、利用可能なユニークな選択可能なマーカーの数によって制限される導入可能なトランス遺伝子の数に実用的な上限がある。一時的な支配的選択(TDS)戦略は、「瘢痕のない」組換えウイルス11の生成を促進することによってこの問題に対処してきた。この戦略を用いて、変異型VACV遺伝子と選択マーカー遺伝子を含むプラスミドをウイルスゲノムに統合するが、追加の隣接するVACV DNAは含まない。このアプローチは、単一のクロスオーバーイベントによる統合の結果として、プラスミド全体の一過性の統合とVACV遺伝子の重複をもたらす。この中間体は、選択圧力下で維持される限り安定であり、この構成体の濃縮を可能にする。選択が除去されると、VACVの複製は、プラスミドの除去と野生型(wt)または組換えウイルスのおよそ50:50比のいずれかで形成される第2のクロスオーバー事象を可能にする。TDSは、外来DNAの安定した導入を必要とせずに組換えウイルスを生成する一方で、複数のウイルスクローンをシーケンシング分析、潜在的に時間とコストのかかるステップによって予想される突然変異をスクリーニングする必要があります。
ここでは、これらのアプローチのそれぞれの最良の側面を組み合わせた組換えポックスウイルスを生成するアプローチを提示し、複製無能修飾ワクシニアアンカラ12、13、1413,14に対12して説明されているアプローチと同様である。この戦略は、視覚的およびホスト範囲の選択を組み合わせて、二重交差事象によって組換えウイルスを迅速に生成し、その後、相同組換えによって選択可能なマーカー遺伝子を排除する。このアプローチは、相同組換えによって媒介される突然変異体の迅速な生成を可能にし、TDSアプローチの「スカーレス」性質を有する一方で、野生型および変異型ウイルスを区別するためのその後のスクリーニングステップを必要としない。我々の方法はまた、抗生物質選択の代わりに宿主範囲選択を使用し、細胞株中の化学的に誘発されたフェノールの変化のリスクを排除する。このアプローチでは、組換えVACVを生成する選択剤として、宿主抗ウイルスタンパク質キナーゼR(PKR)を用いることを選択しました。PKRは、ほとんどの細胞タイプ15において非活性モノマーとして表される。N末端dsRNA結合ドメインで二本鎖RNA(dsRNA)を結合すると、PKRは二量体化し、自己リン酸化された16である。このPKRの活性型は、真核生物開始因子2(eIF2)のαサブユニットをリン酸化し、最終的には開始メチオニルtRNAのリボソームへの送達を阻害し、それによって細胞内翻訳を防止し、多くのウイルスファミリー17,18,18の複製を広く阻害する。
PKRの広範かつ強力な抗ウイルス活性に応答して、多くのウイルスはPKR活性化を防ぐために少なくとも1つの戦略を進化させた。ポックスウイルスの大部分は、2つのPKRアンタゴニストを発現し、VACV中のE3LおよびK3L遺伝子によってコードされ、2つの異なる機構19を介してPKRに拮抗する。E3は、二本鎖RNA20,21と結合することによりPKRホモジマー化を防ぎ、K321は活性化されたPKRに直接結合することにより擬似基質阻害剤として作用し、それによってその基質eIF2α22との相互作用を阻害する。22重要なことに、これら2種のPKRアンタゴニストは、必ずしもすべての種からPKRを阻害するわけではない。例えば、羊痘ウイルスからのK3ホモログは羊由来のPKRを強く阻害し、一方、シープポックスE3ホモログは、かなりのPKR阻害23、24,24を示さなかった。本研究では、蛍光選択と組み合わせたPKR媒介性選択的圧力を用いて、多様な種由来のPKRの有能な細胞では複製できないE3LおよびK3L(VC-R4)に対して削除されたVACV組換え体を生成する方法を提示する。この組換えウイルスは、ネイティブE3Lプロモーターの制御下で遺伝子を発現する組換えウイルスの迅速な生成のための優れた背景を提供します。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. 組み換えベクトルの生成
- 選択カセットを生成するプライマーを設計する。隣接するアンプリコンとベクトルと重なり合う配列を持つ個々のアンプリコンを設計し、いくつかのオンラインプライマー設計ツールのいずれかを使用して、ギブソンアセンブリとも呼ばれるDNA分子の等温酵素アセンブリを容易にします。
注: このプロトコルは、従来の制限エンドヌクレアーゼベースのクローン作成方法を使用して実行することもできます。その場合、重複するシーケンスではなく、適切な制限部位を持つプライマーを設計する。 - ステップ1.1で設計されたプライマーを用いて、PCRは5'から3'まで順に以下の要素を増幅する(図1):E3L(5'アーム)のVACVゲノム領域5'の約500ヌクレオチド、EGFPまたは目的遺伝子、 〜150ヌクレオチドは、E3L(短3'アーム)のVVゲノム領域から直ちに3'、合成早期25/後期ポックスウイルスプロモーター25、mCherry-E3L融合遺伝子、および短い3'腕(長い3'腕)を含むE3LのVACVゲノム領域から〜500ヌクレオチドを含む。
- PCRチューブに、各アンプリコンに対して試薬を次の順序で加える:17μLのDNaseフリー水、各プライマーの1.2μL(初期濃度= 10 μM、 最終濃度=0.5μM、5x PCR反応バッファーの5μL、テンプレートDNA(プラスミドから増幅されたアンプリコン用10 ng:EGFPおよびE/Lプロモーター-mCherry-E3Lカセット;ウイルスゲノムDNAから増幅された増幅素異型の場合は100ng:5'および3'腕)、および0.5μLのDNAポリメラーゼ。50 μLの最終反応量に加えた水の量を調整します。
注:テンプレートDNAの濃度は経験的に決定されるべきであるが、我々は一般的に10 ng /反応から始める。 - チューブをサーモサイクラーに入れ、DNAを98°Cで30sで溶かし、3段階PCRプロトコルの25ラウンド(5sの場合は98°C、10sの場合は55°C、10分間72°C)を使用します。
注: 各プライマー セットのメーカーが推奨する Tm に基づいて、融解温度を決定します。各アンプリコンの長さ(1分/kb)に基づいて、適切な延長時間を決定します。
- PCRチューブに、各アンプリコンに対して試薬を次の順序で加える:17μLのDNaseフリー水、各プライマーの1.2μL(初期濃度= 10 μM、 最終濃度=0.5μM、5x PCR反応バッファーの5μL、テンプレートDNA(プラスミドから増幅されたアンプリコン用10 ng:EGFPおよびE/Lプロモーター-mCherry-E3Lカセット;ウイルスゲノムDNAから増幅された増幅素異型の場合は100ng:5'および3'腕)、および0.5μLのDNAポリメラーゼ。50 μLの最終反応量に加えた水の量を調整します。
- 1%アガロースゲルで増幅産物を可視化します。各DNA製品の10μLと各ウェルに2μLのローディングバッファを加え、8 V /cmで1時間稼働します。
- ゲルはDNAゲル抽出キットおよび製造業者の議定書を使用して各増幅を精製する。50 μLのDNaseフリーウォーターを加え、直ちに遠心分離してカラムから切断機を溶かします。
- EcoRIエンドヌクレアーゼ消化を使用して、pUC19クローニングベクターを直線化します。チューブに、1 μgの pUC19、水を 17 μL、2 L の反応バッファー、および 1 μL (20 単位) のEcoRIを加えます。37°Cで1時間インキュベートする。
- 増幅産物を8V/cmで1時間動かした1%アガロースゲルで可視化し、ステップ1.4で説明したようにDNAゲル抽出キットを使用して製品を精製します。
- マスターミックスキットを使用して、個々のゲル精製アンプリコンおよび線形化ベクターのすべてをリゲートします。
- PCRチューブに、線形化pUC19と各アンプリコン(5'アーム、EGFP、短い3'アーム、E/ Lプロモーター-mCherry-E3Lカセット、3'アーム)の0.2 pmolを加えます。DNaseフリー水を最終体積10μLに加え、10μLのDNAアセンブリマスターミックスを追加します。50°Cで1時間インキュベートする。
- 前述の26、27,27のようにステップ1.6から組み立てられた製品の2μLで化学的に有能な大腸菌を変換します。100 μg/mL アンピシリンを含むLBアガロースプレート上の形質転換細胞のプレート 100 μL。プレートを一晩37°Cでインキュベートする。
- よく隔離されたコロニーを選び、100 μg/mL アンピシリンを含むルリアスープを含む管に個々のコロニーを移す。225rpmで振りながら、37°Cでチューブを一晩インキュベートします。
- プラスミドミニプレップキットを使用して、一晩培養からプラスミドを分離します。分光光度計を用いてDNAの濃度と純度を確認します。A260/A280の比率は1.8~2.0です。
- 希望のクローニング製品が正しいかどうかを判断するために、サンガーシーケンシング用のプラスミドを提出します。DNAを-20°Cに保存します。
2. 組換えウイルスの生成
- 6ウェルプレートで1.0(MOI = 1.0)の感染の多重度で再結合されるウイルスに適した細胞のコンフルエント単層に感染する。感染した細胞を37°Cで、CO2を5%で1時間インキュベートする。その後、培地を吸引し、新鮮なDMEMに置き換えます。感染細胞を37°Cおよび5%CO2でインキュベー2トする。
注:K3L22を欠くワクシニアウイルスのような増殖の有能なウイルスのために、そのようなヨーロッパのウサギの腎臓細胞株RK13(ATCC#CCL-37)またはBSC-40のような細胞株が適している。しかしながら、この論文に記載したウイルスのような複製不十分なウイルスについては、PKRアンタゴニストE3LとK3Lの両方を欠いているが、これら2つの遺伝子をトランスまたはPKRノックダウンまたはノックアウト細胞で発現する補体細胞株が必要である。 - 500 ngのベクターを持つ感染細胞を、製造者のプロトコルに従って市販のトランスフェクション試薬を使用してステップ1.10で生成および検証した。細胞を37°Cで、5%CO2で48時間インキュベートする。2
注:E3LとK3Lの両方を欠くワクシニアウイルスを使用する場合、PKR媒介性選択的圧は、再結合ウイルスの選択を駆動し、これらの細胞内のmCherry-E3L融合タンパク質の発現を維持します。必要に応じて、全体のプラスミドの代わりにトランスフェクションに使用する挿入物のみをPCR増幅することも可能であるべきである。 - 感染後48時間、感染した単層を収穫する。場合によっては、ピペット処理によって細胞を収穫することができますが、まだしっかりと接着している場合は、細胞スクレーパーで収穫します。細胞を3回凍結解凍し、50%振幅で15 sのライセートを超音波処理します。このライセートは-80°Cで使用できる状態になるまで保管してください。
- ステップ2.3で収穫したリセートを10-1から10-6に連続して希釈し、120μLの溶解液をDMEM(10-1)の1080 μLに加え、この希釈液の120μLを1080 μL(10-2)に加え、この4-2回繰り返します。-1各希釈液の1 mLを個々に加え、PKRの有能な細胞株のコンフルエントウェル(この場合はRK13細胞)を加える。
- 感染した細胞を37°Cで、CO2を5%で1時間インキュベートする。その後、培地を吸引し、37°Cおよび5%CO2で感染細胞をインキュベートした新鮮な2DMEMと交換してください。
- 感染後24~48時間、蛍光顕微鏡で組換えウイルスを同定する。組換えウイルス由来のプラークは、mCherry-E3L融合遺伝子の統合による赤色蛍光を発現する(図2)。PKR阻害剤を欠いたウイルスが最初に使用された場合、すべてのプラークは組換えウイルスを含む。
- プラークはRK13細胞上で3回組換えウイルスを精製する。プラークの精製の最終ラウンドの後、すべてのプラークは、赤い蛍光を発現する必要があります。
- VACV PKR阻害剤E3LおよびK3L(RK13+E3L+K3L細胞28)を発現するRK13細胞のコンフルエント6ウェルプレートを、ステップ2.6からプラーク精製赤蛍光ウイルスに感染させる。井戸あたり約50〜100プラークを目指してください。
注:これらの細胞は、トランスでVACV PKR拮抗薬を提供し、mCherry-E3L融合遺伝子を維持するためにPKR媒介性選択的圧力を緩和し、組換えウイルスの「瘢痕のない」生成を促進する。 - EVOS2顕微鏡を用いた蛍光顕微鏡による崩壊したウイルスを同定し、GFPフィルターキューブ(励起:470/22、発光:525/50)およびRFPフィルターキューブ(励起:531/40、放出:593/40)。
注: mCherry-E3L融合遺伝子が失われる頻度は約2.5%です(表2)。EGFPがマーカー遺伝子として含まれていない場合、mCherry-E3L融合遺伝子を失った変異型ウイルスのプラークは無色になります。 - プラークは、RK13+E3L+K3L細胞上で緑色のみの(VC-R4)または無色のプラーク(E3L)を3回精製します。プラークが赤く蛍光を発しないようにしてください。
- pcrおよびサンガーシーケンシングによるmCherry-E3Lの損失および予想される突然変異の存在を確認する。
注:目的の遺伝子または変異がPKR阻害活性を有しない場合、組換えウイルスはRK13+E3L+K3L細胞または同等のPKR阻害またはPKR欠損細胞株上で増殖しなければならない(図3)。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
図1の手順を用いて、PKRアンタゴニストE3LとK3Lの両方を欠いたVACVを生成し、すでにK3Lのために削除されたウイルス中のEGFPにE3Lを置き換えることによって(vP872)。図2は、mCherry-E3Lのウイルス発現を示すPKRの有能RK13細胞における赤色蛍光プラーク、ならびに、mCherry-E3L選択マーカーのE3Lの喪失および崩壊を確認するRK13+E3L+K3L細胞で発現したEGFPを示す。図3は、この組換えウイルス、VC-R4は、PKRの能力のあるRK13細胞において複製できないが、親ウイルスであるvP872E3Lを発現する一方で、複製が有能であることを確認する。RK13細胞内で複製できないことがE3Lの損失によるものであることを確認するために、VC-R4のEGFPをE3Lに置き換え、同じ選択プロトコルを使用して復帰ウイルスを生成しました。図 3では、この復帰ウイルスが RK13 細胞の vP872 と同じ効率で複製されるという検証も行います。興味深いことに、mCherry-E3L選択マーカーの崩壊と一致する無色のプラークは、一般的に「瘢痕のない」組み換え子を選択するために必要なRK13+E3+K3細胞の選択前に同定された。そこで、再結合の効率と崩壊率を決定するために、早期崩壊23の問題を回避するためにポックスウイルスPKRアンタゴニストK3Lを発現するウイルスを生成することを選んだ。図4は、RK13+E3L+K3L細胞の感染後の無色のプラーク(矢印頭)の出現を示す。表1は、3つの独立した実験の結果を示し、その結果、平均12.6%の子孫ビリオンがトランスフェクトされたプラスミドとの再結合を受けたが、以前に報告された周波数29、30、3130,31と同様である。29表2は、RK13+E3L+K3L細胞における全プラークに対する無色プラークの頻度を詳述し、mCherry-E3L選択マーカーの崩壊および喪失率が約1.8%の頻度で発生したことを示す。
図1:p837-GOI-mCherry-E3Lの図と、ホストレンジとビジュアル組み換え戦略(A)5'腕(黒)と3'腕(灰色)はVACVでE3L遺伝子座(茶色)に隣接しています。(B)p837-GOI-mCherry-E3Lにおいて、これらの腕は対象遺伝子(GOI)を含むカセットを横切り、この場合はEGFP(緑色)から分離したmCherry-E3L(赤色)融合遺伝子(赤色)を、短い3'アーム(灰色)で合成初期/後期ポックスウイルスプロモーター25ブルー)に分離した。これらの外的な腕はVACVとp837-GOI-mCherry-E3Lの間の相同の組み換えを促進する。黒い矢印は、ギブソンクローニングによってこのプラスミドを生成するために使用される重複プライマーの部位を示す。(C)PKR選択的圧を除去すると、短い3'腕の間で分子内組換えが行われたウイルスが選択できる。(D) E3L遺伝子に関心のある遺伝子のみを含むウイルス(VC-R4)を生じる。この図の大きなバージョンを表示するには、ここをクリックしてください。

図2:RK13細胞でmCherry(左)とEGFP(右)の両方を発現するp837-GOI-mCherry-E3Lとの組換え24時間後の組換えウイルスプラーク(上)の蛍光顕微鏡写真。(下)組み換えウイルスプラークの顕微鏡写真は、PKR媒介性選択的圧がRK13++細胞で除去されてから48時間後に、EGFP(右)を発現するが、mCherry(左)を発現していない。スケールバーは、すべてのパネルに対して650 μmを示します。この図の大きなバージョンを表示するには、ここをクリックしてください。
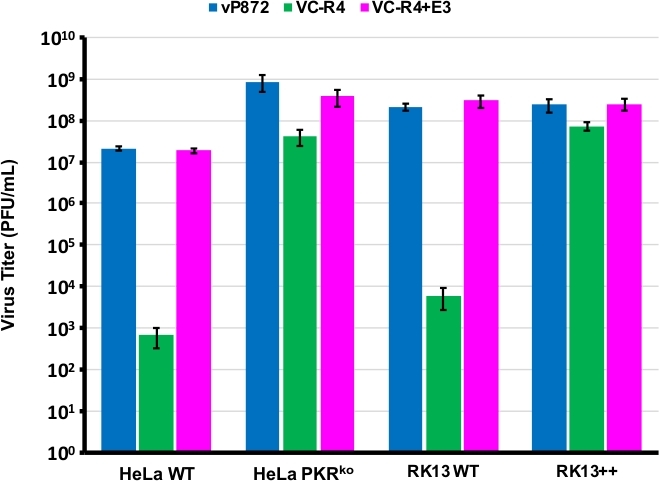
図3:VC-R4は、PKRのコンピテントセルでは複製できません。示された細胞株は、MOI = 0.1でvP872(青)、VC-R4(緑)、またはVC-R4+E3L(マゼンタ)に感染した。感染後48時間、感染細胞を回収し、RK13+E3L+K3L細胞の連続希釈によって刺激した。力乗はPFU/mLで報告され、誤差バーは3回の反復実験の標準偏差を表す。この図の大きなバージョンを表示するには、ここをクリックしてください。

図4 RK13+E3L+K3L細胞におけるmCherry-E3L発現の損失RK13+E3L+K3L細胞に感染したVC-R4+K3L-mCherry-E3Lの蛍光顕微鏡写真と位相コントラスト顕微鏡写真のオーバーレイ。3つのプラークは、VC-R4+K3Lを生み出す選択カセットの崩壊により、もはやmCherry(円)を発現しない。この図の大きなバージョンを表示するには、ここをクリックしてください。
| 実験1 | 実験2 | 実験3 | |
| 赤いプラーク (RK13) | 30 | 11 | 18 |
| 総プラーク(RK13+E3L+K3L) | 225 | 64 | 249 |
| 再結合率 | 13.30% | 17.20% | 7.20% |
表1:P837-K3L-mCherry-E3LプラスミドとのVACVの再結合周波数。
| 実験1 | 実験2 | 実験3 | |
| 総プラーク(RK13+E3L+K3L) | 115 | 44 | 210 |
| 無色プラーク(RK13+E3L+K3L) | 3 | 1 | 1 |
| 再結合率 | 2.60% | 2.30% | 0.50% |
表2:RK13+E3+K3細胞におけるVC-R4+K3L-mCherry-E3LからのmCherry-E3L損失の頻度。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
ここでは、最終組換えウイルスに外来DNAを保持せずに組換えワクシニアウイルスを生成する過渡マーカー選択戦略32のバリエーションを提示する。我々の戦略は、抗生物質などの選択的圧力の他の形態ではなく、宿主抗ウイルスタンパク質PKRによって媒介される選択的圧力を使用する。宿主抗ウイルス遺伝子の使用は、細胞内の化学的に誘発された表型変化の可能性、または選択薬物による突然変異のリスクの増加を排除する。さらに、薬物選択とは異なり、PKRはすべての細胞で構成的に発現されるため、我々のアプローチにはラグフェーズがない。また、mCherry発現に基づく二次的な視覚選択は、第1段階でトランスジーンを発現するプラークのみがピックされ、mCherry-E3L遺伝子を失った成熟した組換えウイルスを選択する際に陰性選択的マーカーとして同様に効率的であることを保証することにより、この方法の特異性を向上させる。
この組換え戦略の最も重要なステップは、適切な組換えベクターの生成、および選択されたウイルスがクローン性であることを保証するための適切なプラーク精製です。本論文では、組換えベクトルを生成する「ギブソンアセンブリ」を提案する。この戦略は非常に効率的であり、1日で再結合ベクターを含むすべての断片の組み立てを可能にする。しかし、短い3'腕と長い3'腕は同一の配列を共有するので、これらの断片はクローニング反応の間に結合される可能性があり、一部のベクトルはmCherry-E3Lカセットを含まないかもしれません。私たちの経験では、これはまれですが、クローニング後のベクターの構造を確認する必要があります。我々はまた、従来のエンドヌクレアーゼおよびリガーゼ法を用いて、この戦略のための組換えベクターを生成した。この戦略は、上記の問題を回避しますが、より多くの労力を要する可能性があります。プラーク精製は一般的に簡単であり、主に最初の再結合に適切な寛容細胞を使用することに依存しており、組換えウイルスのみが複製できるように初期プラーク精製のためのPKRコンピテント細胞、そして再び寛容な細胞を使用して分子内組換えおよび選択可能マーカーの喪失を促進する。したがって、この戦略を効果的かつ効果的に適用するには、セルラインに注意が必要です。
本研究では、この方法を用いて、PKR拮抗薬E3LおよびK3Lの両方について削除されたVACV組換え遺伝子を生成し、E3Lプロモーターの制御下でEGFPを発現させる方法を示す。今後、このウイルスは、PKRの有能な細胞で複製することができないため、将来の組換えウイルスの効率的な背景として機能します。したがって、mCherry-E3L組換えカセットを子孫のビリオンに駆動すると同時に、本質的に非組換えウイルスの複製を防止するための強力なPKR媒介選択的圧力が存在する。さらに、組換えカセットの取り込みによるEGFPの喪失は、選ばれたプラークが非組換えウイルスと共感染していないことを保証するのに有用な二次選択マーカーである。これまで報告されたVACVの速度と一致する再結合率を観察したが、視覚蛍光マーカーは、適切な組換えウイルスが選択される可能性を高めることで、組換えウイルスの生成効率を高める。PKRの有能な細胞上の選択の2ラウンド後の無色プラークの観察は、おそらくE3LとmCherry-E3Lマーカー遺伝子の間の同一配列の長さの増加のために、mCherry-E3L損失の速度が3'短い腕の長さを増減することによって「調整」される可能性があることを示唆している。この技術の主な制限は、組換え剤の選択的圧力としてPKRを使用することである。この組換え戦略の最も効率的な使用は、PKRアンタゴニストを欠いているバックグラウンドでこれらのウイルスを生成することです。しかし、この着色選択マーカーは、単にmCherry発現プラークを精製するプラークによって、PKRによって媒介された選択なしでも、この組み換え戦略を使用することを可能にする。PKR媒介性選択的圧力の欠如は、最初のスクリーニングステップの効率を低下させるが、mCherry発現プラークの割合は依然として色ベースの選択が実行可能であるほど高い。したがって、この方法は、ポックスウイルスゲノムにほぼすべての遺伝子を挿入するために使用することができる。
EGFPの挿入によって示されるように、このアプローチにより、任意の遺伝子を、ネイティブプロモーターの制御下にあるE3L遺伝子に迅速に挿入することができ、トランスジーンがPKRアンタゴニストでない場合にはPKRヌル細胞または補完細胞株が下流実験に使用されることを願う。この戦略は、ここで報告するVC-R4ウイルスと組み合わせることで、プロセスの早い段階でホスト媒介性選択的圧力と組換え遺伝子の視覚的同定を使用して、組み換えワクシニアウイルスを迅速かつ確実に生成する新しい強力な方法を追加します。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者らは、競合する財政的利益を宣言しない。
Acknowledgments
このプロジェクトは、国立衛生研究所(AI114851)がSRに資金を提供しました。
Materials
| Name | Company | Catalog Number | Comments |
| 2X-Q5 Master Mix | NEB | M0492L | High-fidelity polymerase used in PCR |
| Ampicillin | ThermoFisher Scientific | 11593027 | Bacterial selective agent |
| Disposable Cell Scrapers | ThermoFisher Scientific | 08-100-242 | Cell scraper to harvest infected cells |
| EVOS FL Auto 2 Cell imaging system | ThermoFisher Scientific | AMAFD2000 | Fluorescent microscope |
| EVOS Light Cube, GFP | ThermoFisher | AMEP4651 | GFP Cube |
| EVOS Light Cube, RFP | ThermoFisher | AMEP4652 | RFP Cube |
| GenJet | SignaGen Laboratories | SL100489 | Transfection reagent |
| Luria Bertani (LB) Broth | Gibco | 10855021 | Bacterial growth medium |
| Monarch DNA gel extraction kit | NEB | T1020L | Gel purification kit used to purify amplicons and linearized vectors |
| Monarch Plasmid Miniprep kit | NEB | T1010L | Miniprep kit ussed to purify plasmids |
| NanoDrop One | ThermoFisher Scientific | ND-ONE-W | Spectrophotometer used to measure RNA and DNA concentration |
| NEBuilder Master Mix | NEB | E2621L | Isothermal enzymatic assembly kit used to generate the recombination vector |
| Q500 Sonicator | Qsonica | Q500-110 | Sonicator for virus lysates |
| RK13 cells | ATCC | CCL-37 | Rabbit kidney cells |
| VWR Multiwell Cell Culture plates | VWR | 10062-892 | Cell culture plates |
References
- Brochier, B., et al. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature. 354 (6354), 520-522 (1991).
- Pastoret, P. P., Brochier, B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; a link between Jenner and Pasteur. Epidemiology and Infection. 116 (3), 235-240 (1996).
- Chan, W. M., McFadden, G.
- Nguyen, D. H., et al. Vaccinia virus-mediated expression of human erythropoietin in tumors enhances virotherapy and alleviates cancer-related anemia in mice. Molecular Therapy. 21 (11), 2054-2062 (2013).
- Frentzen, A., et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proceedings of the National Academy of Sciences of the United States of America. 106 (31), 12915-12920 (2009).
- Pastoret, P. P., Vanderplasschen, A.
- COLLIER, L. H. The development of a stable smallpox vaccine. The Journal of Hygiene. 53 (1), 76-101 (1955).
- Weir, J. P., Bajszár, G., Moss, B. Mapping of the vaccinia virus thymidine kinase gene by marker rescue and by cell-free translation of selected mRNA. Proceedings of the National Academy of Sciences of the United States of America. 79 (4), 1210-1214 (1982).
- Mackett, M., Smith, G. L., Moss, B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proceedings of the National Academy of Sciences of the United States of America. 79 (23), 7415-7419 (1982).
- Nakano, E., Panicali, D., Paoletti, E. Molecular genetics of vaccinia virus: demonstration of marker rescue. Proceedings of the National Academy of Sciences of the United States of America. 79 (5), 1593-1596 (1982).
- Falkner, F. G., Moss, B. Transient dominant selection of recombinant vaccinia viruses. Journal of Virology. 64 (6), 3108-3111 (1990).
- Staib, C., Drexler, I., Ohlmann, M., Wintersperger, S., Erfle, V., Sutter, G. Transient Host Range Selection for Genetic Engineering of Modified Vaccinia Virus Ankara. BioTechniques. 28 (6), 1137-1148 (2000).
- Staib, C., Drexler, I., Sutter, G. Construction and Isolation of Recombinant MVA. Vaccinia Virus and Poxvirology. , 77-99 (2004).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Pfaller, C. K., Li, Z., George, C. X., Samuel, C. E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Current Opinion in Immunology. 23 (5), 573-582 (2011).
- Bevilacqua, P. C., George, C. X., Samuel, C. E., Cech, T. R. Binding of the protein kinase PKR to RNAs with secondary structure defects: Role of the tandem A - G mismatch and noncontigous helixes. Biochemistry. 37 (18), 6303-6316 (1998).
- Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., Hinnebusch, A. G. Tight Binding of the Phosphorylated Subunit of Initiation Factor 2 (eIF2) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Molecular and Cellular Biology. 21 (15), 5018-5030 (2001).
- Rothenburg, S., Georgiadis, M. M., Wek, R. C. Evolution of eIF2α kinases: Adapting translational control to diverse stresses. Evolution of the Protein Synthesis Machinery and Its Regulation. , 235-260 (2016).
- Bratke, K. A., McLysaght, A., Rothenburg, S. A survey of host range genes in poxvirus genomes. Infection, Genetics and Evolution. 14, 406-425 (2013).
- Chang, H. W., Watson, J. C., Jacobs, B. L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proceedings of the National Academy of Sciences. 89 (11), 4825-4829 (1992).
- Romano, P. R., et al. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Molecular and Cellular Biology. 18 (12), 7304-7316 (1998).
- Beattie, E., Tartaglia, J., Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 183 (1), 419-422 (1991).
- Park, C., Peng, C., Brennan, G., Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Annals of the New York Academy of Sciences. 1438 (1), 18-29 (2019).
- Rothenburg, S., Brennan, G. Species-Specific Host-Virus Interactions: Implications for Viral Host Range and Virulence. Trends in Microbiology. , (2019).
- Chakrabarti, S., Sisler, J. R., Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 23 (6), 1094-1097 (1997).
- Chung, C. T., Niemela, S. L., Miller, R. H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution (recombinant DNA). Biochemistry. 86, 2172-2175 (1989).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Methods in Enzymology. 218, 621-627 (1993).
- Rahman, M. M., Liu, J., Chan, W. M., Rothenburg, S., McFadden, G. Myxoma Virus Protein M029 Is a Dual Function Immunomodulator that Inhibits PKR and Also Conscripts RHA/DHX9 to Promote Expanded Host Tropism and Viral Replication. PLOS Pathogens. 9 (7), 1003465 (2013).
- Evans, D. H., Stuart, D., McFadden, G. High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus-infected mammalian cells. Journal of Virology. 62 (2), 367-375 (1988).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. Journal of Virology. 61 (6), 1788-1795 (1987).
- Spyropoulos, D. D., Roberts, B. E., Panicali, D. L., Cohen, L. K. Delineation of the viral products of recombination in vaccinia virus-infected cells. Journal of Virology. 62 (3), 1046-1054 (1988).
- Liu, L., et al. Transient dominant host-range selection using Chinese hamster ovary cells to generate marker-free recombinant viral vectors from vaccinia virus. BioTechniques. 62 (4), 183-187 (2017).
