La maggior parte delle sinapsi eccitatorie nel sistema nervoso centrale si trovano su spine dendritiche – piccole sporgenze di una membrana neuronale. Queste sporgenze formano compartimenti biochimici confinati che controllano la trasduzione del segnale intracellulare. La plasticità strutturale delle spine dendritiche e delle sinapsi è strettamente correlata ai cambiamenti funzionali nell’efficacia sinaptica che sono alla base di processi importanti come l’apprendimento e la memoria1,2. È importante notare che la microscopia elettronica (EM) è l’unica tecnica che consente di determinare se una colonna vertebrale dendritica ha un input presinaptico. La risoluzione EM è necessaria anche per studiare dettagli ultrastrutturali come la forma di una densità postsinaptica (PSD), che rappresenta una parte postsinaptica di una sinapsi, o le dimensioni di una colonna vertebrale dendritica, nonché le dimensioni e la forma di un bouton assonale. Inoltre, con EM è possibile visualizzare le sinapsi e l’ambiente circostante.
Grazie ai progressi delle tecnologie di imaging e calcolo è possibile ricostruire interi circuiti neurali. Le tecniche di microscopia elettronica a volume, come la microscopia elettronica a trasmissione a sezione seriale (ssTEM), la microscopia elettronica a scansione a blocchi seriali (SBEM) e la microscopia elettronica a scansione a fascio ionico focalizzato (FIB-SEM) sono comunemente utilizzate per le ricostruzioni di circuiti neuronali3.
Nei nostri studi, il metodo SBEM è impiegato con successo per studiare la plasticità strutturale delle spine dendritiche e delle PSD in campioni di ippocampo di topo e fette di cervello organotipico 4,5. Lo SBEM si basa sull’installazione di un ultramicrotomo in miniatura all’interno della camera del microscopio elettronico a scansione6,7,8,9. La parte superiore del blocco campione viene tagliata, quindi il campione viene tagliato a una profondità specificata dall’ultramicrotomo, rivelando una nuova faccia del blocco, che viene nuovamente tagliata e quindi il processo viene ripetuto8. Di conseguenza, rimane solo l’immagine di una faccia di blocco mentre la fetta che è stata tagliata viene persa come detriti. Ecco perché SBEM è chiamata una tecnica distruttiva, il che significa che non è possibile immaginare di nuovo lo stesso posto. Tuttavia, il vantaggio dei metodi distruttivi on-block è che non soffrono di problemi di deformazione e perdita di sezione che possono influire in modo significativo sulla qualità dei dati e sull’analisi dei dati3. Inoltre, SBEM dà la possibilità di visualizzare un campo visivo relativamente ampio (> 0,5 mm × 0,5 mm) ad alta risoluzione3.
Per utilizzare SBEM, i campioni devono essere preparati secondo un protocollo dedicato e altamente contrastante a causa del rilevatore di elettroni retrodatterato utilizzato per l’acquisizione di immagini. Mostriamo qui come eseguire la preparazione del campione secondo il protocollo basato su una procedura sviluppata da Deerinck10 (National Center for Microscopy and Imaging Research (NCMIR) metodo), utilizzando macchie ridotte di osmio-tiocarboidrazide-osmio (rOTO) sviluppate nel 19808,11. Inoltre, introduciamo un approccio di fissazione in due fasi, con lieve perfusione di fissazione che consente di utilizzare lo stesso cervello sia per studi di immunofluorescenza con microscopia ottica e SBEM.
Nel protocollo un cervello di topo viene fissato principalmente con un lieve fissativo, e poi tagliato a metà, e un emisfero viene postfisso e preparato per l’immunofluorescenza (IF), mentre l’altro per gli studi EM (Figura 1).

Figura 1. Rappresentazione schematica del flusso di lavoro per la preparazione delle spine dendritiche per l’analisi con SBEM. I topi sono stati sacrificati e perfusi con un lieve fissativo primario. Il cervello è stato tagliato a metà e un emisfero è stato postfisso con fissativo dedicato all’immunofluorescenza (IF), crioprotetto, affettato usando un criostato ed elaborato per studi IF, mentre l’altro emisfero è stato postfisso con fissativo EM, affettato con il vibratoma e preparato per gli studi EM. Le fette di cervello per gli studi SBEM sono state contrastate, piatte incorporate nella resina, quindi una regione CA1 dell’ippocampo è stata montata sul perno e tagliata con SBEM (Figura 1). La parte del protocollo evidenziata in una casella gialla è stata presentata nel video. Fare clic qui per visualizzare una versione più grande di questa figura.
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 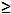 98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands |
50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands |
10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
