La mayoría de las sinapsis excitatorias en el sistema nervioso central se encuentran en las espinas dendríticas, pequeñas protuberancias de una membrana neuronal. Estas protuberancias forman compartimentos bioquímicos confinados que controlan la transducción de señales intracelulares. La plasticidad estructural de las espinas dendríticas y las sinapsis está estrechamente relacionada con los cambios funcionales en la eficacia sináptica que subyacen a procesos tan importantes como el aprendizaje y la memoria1,2. Es importante tener en cuenta que la microscopía electrónica (EM) es la única técnica que permite determinar si una columna dendrítica tiene una entrada presináptica. La resolución EM también es necesaria para estudiar detalles ultraestructurales como la forma de una densidad postsináptica (PSD), que representa una parte postsináptica de una sinapsis, o las dimensiones de una columna dendrítica, así como el tamaño y la forma de un bouton axonal. Además, con EM es posible visualizar las sinapsis y su entorno.
Gracias a los avances en las tecnologías de imagen y computación es posible reconstruir circuitos neuronales completos. Las técnicas de microscopía electrónica de volumen, como la microscopía electrónica de transmisión de sección serie (ssTEM), la microscopía electrónica de barrido de cara de bloque serie (SBEM) y la microscopía electrónica de barrido de haz de iones enfocado (FIB-SEM) se utilizan comúnmente para reconstrucciones de circuitos neuronales3.
En nuestros estudios, el método SBEM se emplea con éxito para investigar la plasticidad estructural de las espinas dendríticas y las PSD en muestras del hipocampo del ratón y cortes cerebrales organotípicos 4,5. El SBEM se basa en la instalación de un ultramicrotomo en miniatura dentro de la cámara del microscopio electrónico de barrido6,7,8,9. Se toma una imagen de la parte superior del bloque de muestra, y luego el ultramicrotomo corta la muestra a una profundidad especificada, revelando una nueva cara de bloque, que se vuelve a obtener una imagen y luego se repite el proceso8. Como resultado, solo queda la imagen de una cara de bloque, mientras que la rebanada que se ha cortado se pierde como escombros. Es por eso que SBEM se llama una técnica destructiva, lo que significa que no es posible obtener imágenes del mismo lugar nuevamente. Sin embargo, la ventaja de los métodos destructivos en bloque es que no sufren problemas de deformación y pérdida de sección que pueden afectar significativamente la calidad de los datos y el análisis de datos3. Además, SBEM ofrece la posibilidad de obtener imágenes de un campo de visión relativamente grande (> 0,5 mm × 0,5 mm) a alta resolución3.
Para emplear SBEM, las muestras deben prepararse de acuerdo con un protocolo dedicado y altamente contrastante debido al detector de electrones retrodibuerados utilizado para adquirir imágenes. Mostramos aquí cómo realizar la preparación de muestras de acuerdo con el protocolo basado en un procedimiento desarrollado por Deerinck10 (método del Centro Nacional de Microscopía e Investigación de Imágenes (NCMIR)), utilizando tinciones reducidas de osmio-tiocarbohidrazida-osmio (rOTO) desarrolladas en la década de 19808,11. Además, introducimos un enfoque de fijación en dos pasos, con perfusión de fijación leve que permite utilizar el mismo cerebro tanto para estudios de inmunofluorescencia con microscopía de luz como para SBEM.
En el protocolo, un cerebro de ratón se fija principalmente con un fijador suave, y luego se corta en mitades, y un hemisferio se posfija y se prepara para la inmunofluorescencia (IF), mientras que el otro para los estudios EM(Figura 1).

Figura 1. Representación esquemática del flujo de trabajo para la preparación de espinas dendríticas para el análisis con SBEM. Los ratones fueron sacrificados y perfundidos con un fijador primario suave. El cerebro se cortó en mitades, y un hemisferio se colocó con fijador dedicado a la inmunofluorescencia (IF), crioprotegido, se cortó en rodajas con un criostato y se procesó para estudios de FI, mientras que el otro hemisferio se posfijó con fijador EM, se cortó con el vibrátomo y se preparó para los estudios em. Las rebanadas cerebrales para los estudios de SBEM se contrastaron, se incrustaron de forma plana en resina, luego se montó una región CA1 del hipocampo en el pasador y se tomaron imágenes con SBEM(Figura 1). La parte del protocolo resaltada en un cuadro amarillo se presentó en el video. Haga clic aquí para ver una versión más grande de esta figura.
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 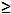 98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands |
50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands |
10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
