

Summary
يوفر البروتوكول الموضح هنا تعليمات مفصلة حول كيفية تحليل المناطق الجينومية ذات الأهمية لإمكانات ترميز البروتين الدقيق باستخدام PhyloCSF على متصفح UCSC Genome سهل الاستخدام. بالإضافة إلى ذلك ، يوصى بالعديد من الأدوات والموارد لمواصلة التحقيق في خصائص تسلسل البروتينات الدقيقة المحددة لاكتساب نظرة ثاقبة على وظائفها المفترضة.
Abstract
دفع الجيل التالي من التسلسل (NGS) مجال علم الجينوم إلى الأمام وأنتج تسلسلات جينوم كاملة للعديد من الأنواع الحيوانية والكائنات الحية النموذجية. ومع ذلك ، على الرغم من هذه الثروة من معلومات التسلسل ، فقد أثبتت جهود التعليق التوضيحي الشامل للجينات أنها تمثل تحديا ، خاصة بالنسبة للبروتينات الصغيرة. والجدير بالذكر أن طرق التعليق التوضيحي التقليدية للبروتين تم تصميمها لاستبعاد البروتينات المفترضة المشفرة بواسطة إطارات قراءة مفتوحة قصيرة (sORFs) يقل طولها عن 300 نيوكليوتيدات لتصفية العدد الأكبر أضعافا مضاعفة من sORFs الزائفة غير المشفرة في جميع أنحاء الجينوم. ونتيجة لذلك ، تم تصنيف مئات البروتينات الصغيرة الوظيفية التي تسمى البروتينات الدقيقة (<100 حمض أميني في الطول) بشكل غير صحيح على أنها الحمض النووي الريبي غير المشفر أو تم تجاهلها تماما.
نقدم هنا بروتوكولا مفصلا للاستفادة من أدوات المعلوماتية الحيوية المجانية والمتاحة للجمهور للاستعلام عن المناطق الجينومية لإمكانات ترميز البروتين الدقيق بناء على الحفظ التطوري. على وجه التحديد ، نقدم إرشادات خطوة بخطوة حول كيفية فحص إمكانات حفظ التسلسل والترميز باستخدام ترددات استبدال الكودون الوراثي (PhyloCSF) على متصفح الجينوم بجامعة كاليفورنيا سانتا كروز (UCSC) سهل الاستخدام. بالإضافة إلى ذلك ، نقوم بتفصيل الخطوات اللازمة لتوليد محاذاة أنواع متعددة بكفاءة من تسلسلات البروتين الدقيق المحددة لتصور الحفاظ على تسلسل الأحماض الأمينية والتوصية بالموارد اللازمة لتحليل خصائص البروتين الدقيق ، بما في ذلك هياكل المجال المتوقعة. يمكن استخدام هذه الأدوات القوية للمساعدة في تحديد تسلسلات ترميز البروتين الدقيق المفترضة في المناطق الجينومية غير القانونية أو لاستبعاد وجود تسلسل ترميز محفوظ مع إمكانات انتقالية في نسخة غير مشفرة ذات أهمية.
Introduction
كان تحديد المجموعة الكاملة من عناصر الترميز في الجينوم هدفا رئيسيا منذ بدء مشروع الجينوم البشري ، ولا يزال هدفا مركزيا نحو فهم النظم البيولوجية ومسببات الأمراض القائمة على الوراثة1،2،3،4. أدى التقدم في تقنيات NGS إلى إنتاج تسلسلات جينوم كاملة لعدد كبير من الكائنات الحية ، بما في ذلك الفقاريات واللافقاريات والخميرة والنباتات5. بالإضافة إلى ذلك ، كشفت طرق التسلسل النسخي عالية الإنتاجية عن تعقيد النسخ الخلوي ، وحددت الآلاف من جزيئات الحمض النووي الريبي الجديدة مع كل من وظائف ترميز البروتين وغير المشفرة 6,7. يعد فك تشفير هذا الكم الهائل من معلومات التسلسل عملية مستمرة ، ولا تزال هناك تحديات مع جهود التعليق التوضيحي الجيني الشاملة8.
وقد قدم التطور الأخير لأساليب التنميط الانتقالي، بما في ذلك التنميط الريبوسومي9,10 وتسلسل الريبوسوم المتعدد 11، أدلة تشير إلى أن المئات من أحداث الترجمة غير القانونية ترسم خريطة ل sORFs غير المشروحة حاليا في جميع أنحاء الجينوم، مع إمكانية توليد بروتينات صغيرة تسمى البروتينات الدقيقة أو الببتيدات الدقيقة12,13,14,15,16، 17. ظهرت البروتينات الدقيقة كفئة جديدة من البروتينات متعددة الاستخدامات التي تم تجاهلها سابقا بطرق التعليق التوضيحي الجيني القياسية بسبب صغر حجمها (<100 حمض أميني) وعدم وجود خصائص جينية كلاسيكية مشفرة للبروتين8،12،18،19،20. تم وصف البروتينات الدقيقة في جميع الكائنات الحية تقريبا ، بما في ذلك الخميرة 21،22 ، والذباب 17،23،24 ، والثدييات25،26،27،28 ، وقد ثبت أنها تلعب أدوارا حاسمة في عمليات متنوعة ، بما في ذلك التنمية والتمثيل الغذائي وإشارات الإجهاد19،20،29 ، 30,31,32,33,34. وبالتالي ، من الضروري الاستمرار في تعدين الجينوم لأعضاء إضافيين من هذه الفئة التي تم تجاهلها منذ فترة طويلة من البروتينات الصغيرة الوظيفية.
وعلى الرغم من الاعتراف الواسع النطاق بالأهمية البيولوجية للبروتينات الدقيقة، لا تزال هذه الفئة من الجينات ممثلة تمثيلا ناقصا إلى حد كبير في شروح الجينوم، ولا يزال تحديدها الدقيق يمثل تحديا مستمرا أعاق التقدم في هذا المجال. تم مؤخرا تطوير العديد من الأدوات الحسابية والأساليب التجريبية للتغلب على الصعوبات المرتبطة بتحديد تسلسلات ترميز البروتين الدقيق (نوقشت على نطاق واسع في العديد من المراجعات الشاملة8،35،36،37). اعتمدت العديد من دراسات تحديد البروتين الدقيق الحديثة 38,39,40,41,42,43,44,45,46,47 بشكل كبير على استخدام خوارزمية واحدة تسمى PhyloCSF 48,49 ، وهو نهج قوي في علم الجينوم المقارن يمكن الاستفادة منه للتمييز بين مناطق ترميز البروتين المحفوظة في الجينوم وتلك التي لا ترميزها.
يقارن PhyloCSF ترددات استبدال الكودون (CSF) باستخدام محاذاة النيوكليوتيدات متعددة الأنواع والنماذج الجينية للكشف عن البصمات التطورية للجينات المشفرة للبروتين. يعتمد هذا النهج التجريبي القائم على النموذج على فرضية أن البروتينات محفوظة في المقام الأول على مستوى الأحماض الأمينية بدلا من تسلسل النيوكليوتيدات. لذلك ، يتم تسجيل بدائل الكودون المترادفة ، التي تشفر نفس الحمض الأميني ، أو بدائل الكودون للأحماض الأمينية ذات الخصائص المحفوظة (أي الشحنة ، كره الماء ، القطبية) بشكل إيجابي ، في حين أن البدائل غير المترادفة ، بما في ذلك البدائل الخاطئة والهراء ، تسجل بشكل سلبي. تم تدريب PhyloCSF على بيانات الجينوم الكامل وأثبت فعاليته في تسجيل أجزاء قصيرة من تسلسل الترميز (CDS) بمعزل عن التسلسل الكامل ، وهو أمر ضروري عند تحليل البروتينات الدقيقة أو الإكسونات الفردية لجينات ترميز البروتين القياسية48,49.
ومن الجدير بالذكر أن التكامل الأخير لمحاور المسار PhyloCSF في متصفح الجينوم 49,50,51 بجامعة كاليفورنيا سانتا كروز (UCSC) يمكن الباحثين من جميع الخلفيات من الوصول بسهولة إلى واجهة سهلة الاستخدام للاستعلام عن المناطق الجينومية ذات الأهمية لإمكانات ترميز البروتين. يوفر البروتوكول الموضح أدناه تعليمات مفصلة حول كيفية تحميل محاور تتبع PhyloCSF على متصفح UCSC Genome Browser ومن ثم استجواب المناطق الجينومية ذات الأهمية للتحقيق في مناطق ترميز البروتين عالية الثقة (أو عدم وجودها). بالإضافة إلى ذلك ، في حالة ملاحظة درجة PhyloCSF إيجابية ، يتم تحديد الخطوات لمواصلة تحليل إمكانات ترميز البروتين الدقيق وتوليد محاذاة أنواع متعددة بكفاءة من تسلسل الأحماض الأمينية المحددة لتوضيح الحفاظ على تسلسل الأنواع المتقاطعة. وأخيرا، يتم إدخال العديد من الموارد والأدوات الإضافية المتاحة للجمهور في المناقشة لمسح خصائص البروتين الدقيق المحددة، بما في ذلك هياكل المجال المتوقعة والنظرة الثاقبة لوظيفة البروتين الدقيق المفترضة.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
يوضح البروتوكول الموضح أدناه تفاصيل خطوات تحميل مسارات متصفح PhyloCSF والتنقل فيها على متصفح UCSC Genome Browser (الذي تم إنشاؤه بواسطة Mudge et al.49). للأسئلة العامة المتعلقة بمتصفح UCSC Genome ، يمكن العثور على دليل مستخدم متصفح الجينوم الشامل هنا: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. تحميل مركز تتبع PhyloCSF إلى متصفح الجينوم UCSC
- افتح نافذة متصفح الإنترنت وانتقل إلى متصفح UCSC Genome Browser (https://genome.ucsc.edu/).
- ضمن عنوان أدواتنا ، حدد الخيار تتبع المحاور .
ملاحظة: يمكن أيضا العثور على خيار تعقب المحاور ضمن علامة التبويب بياناتي . - في علامة التبويب مراكز الوصل العامة ، اكتب PhyloCSF في المربع مصطلحات البحث . انقر فوق الزر البحث في المحاور العامة .
- اتصل ب PhyloCSF بالنقر فوق الزر " اتصال" لاسم المحور PhyloCSF (الوصف: إمكانات ترميز البروتين التطوري كما تم قياسها بواسطة PhyloCSF ).
ملاحظة: سيتم تحميل محور المسار هذا إلى العديد من التجميعات، بما في ذلك الإنسان (hg19 وhg38) والماوس (mm10 وmm39). - بعد النقر فوق اتصال، انتظر حتى تتم إعادة توجيهك إلى صفحة بوابة متصفح الجينوم UCSC (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. الانتقال إلى الجينات ذات الأهمية باستخدام معرفات الجينات
- حدد الأنواع وتجميع الجينوم للاستعلام عنه. للاستعلام عن نوع مختلف (على سبيل المثال، الماوس)، حدد الأنواع ذات الأهمية ضمن عنوان تصفح/تحديد الأنواع بالنقر فوق الرمز المناسب، أو اكتب الأنواع في مربع النص الذي يقول، أدخل الأنواع أو الاسم الشائع أو معرف التجميع.
ملاحظة: يتم سرد التجميع مباشرة تحت عنوان البحث عن موضع . عادة ما يكون الإعداد الافتراضي هو الجمعية البشرية (على سبيل المثال ، ديسمبر 2009 [GRCh37/hg19]). - اختر التجميع للبحث تحت عنوان البحث عن موضع باستخدام القائمة المنسدلة.
- أدخل الموضع أو رمز الجين أو مصطلحات البحث في مربع الموضع / مصطلح البحث وانقر على Go للانتقال إلى جين ذي أهمية على متصفح الجينوم.
- إذا أسفر البحث عن تطابقات متعددة، فانتظر حتى تتم إعادة توجيهك إلى صفحة تتطلب تحديد موضع اهتمام. انقر على الجين المناسب محل الاهتمام.
3. الانتقال إلى المناطق الجينومية ذات الأهمية باستخدام معلومات التسلسل
- انتقل إلى متصفح UCSC Genome Browser (https://genome.ucsc.edu/) وحدد أداة المحاذاة الشبيهة بالانفجار (BLAT) ضمن عنوان أدواتنا للاستعلام عن تسلسل معين من الحمض النووي أو البروتين. بدلا من ذلك، مرر مؤشر الماوس فوق علامة التبويب أدوات وحدد الخيار بلات أو اتبع هذا الرابط: https://genome.ucsc.edu/cgi-bin/hgBlat.
- حدد الأنواع (الجينوم) وتجميع الاهتمام باستخدام القوائم المنسدلة.
- حدد نوع الاستعلام باستخدام القائمة المنسدلة.
- الصق تسلسل الاهتمام في مربع النص BLAT Search Genome وانقر فوق إرسال.
- انقر على رابط المتصفح تحت عنوان ACTIONS للانتقال إلى المنطقة الجينومية ذات الاهتمام.
4. تحديد sORFs المحفوظة باستخدام بيانات تتبع PhyloCSF
- مسح بصري لمنطقة الاهتمام الجينومية لتسجيل مناطق PhyloCSF بشكل إيجابي (الشكل 1).
ملاحظة: للحصول على شرح مفصل لكيفية تفسير درجات PhyloCSF بصريا على متصفح UCSC Genome ، راجع قسم النتائج التمثيلية أدناه. - استخدم ميزة التكبير/التصغير لتكبير المناطق ذات الأهمية لفحص خصائص التسلسل والبحث عن كودونات البدء/الإيقاف. للتكبير يدويا، اضغط مع الاستمرار على مفتاح shift وانقر مع الاستمرار فوق زر الماوس أثناء السحب على طول المنطقة محل الاهتمام. بدلا من ذلك، استخدم زري التكبير والتصغير في أعلى الصفحة للتنقل (تتوفر خيارات التكبير/التصغير 1.5x أو 3x أو 10x أو base).
ملاحظة: قبل استخدام أزرار التكبير/التصغير ، من الضروري تغيير موضع الجين بحيث تكون المنطقة محل الاهتمام في منتصف الشاشة. لتنفيذ هذا الإجراء ، انقر فوق الصورة واسحبها إلى اليسار أو اليمين لتحريك المنطقة الجينومية أفقيا حسب الرغبة أو استخدم أسهم الحركة في أعلى الصفحة. - قم بالتكبير حتى يصبح تسلسل النيوكليوتيدات (القاعدة) مرئيا.
ملاحظة: سيظهر تسلسل النيوكليوتيدات مباشرة فوق درجة +1 PhyloCSF الملساء. - مسح بصري لتسلسل النيوكليوتيدات بالقرب من بداية ونهاية مناطق PhyloCSF ذات الدرجات الإيجابية لتحديد كودونات البداية المفترضة (ATG) والتوقف (TGA / TAA / TAG).
ملاحظة: إذا كان الجين محل الاهتمام موجودا على الشريط الناقص من الحمض النووي، فإن كودونات البدء والتوقف ستكون المكمل العكسي (أي CAT لكودون البدء وTCA/TTA/CTA لكودون التوقف).
5. عرض المناطق المتجانسة في الجينومات الأخرى
- مرر مؤشر الماوس فوق عنوان عرض في أعلى الصفحة وانقر على الخيار في الجينومات الأخرى (تحويل ).
- حدد الجينوم محل الاهتمام باستخدام القائمة المنسدلة أسفل عنوان الجينوم الجديد .
- حدد التجميع الجينومي الذي يهمه باستخدام القائمة المنسدلة أسفل عنوان " تجميع جديد "، ثم انقر على الزر "إرسال ".
- بمجرد أن يقوم المتصفح بإرجاع قائمة بالمناطق في التجميع الجديد مع التشابه ، انقر فوق رابط موضع الكروموسوم للانتقال إلى المنطقة المتماثلة ذات الاهتمام.
ملاحظة: سيتم تحديد النسبة المئوية لمجموع القواعد (النيوكليوتيدات) والمدى الذي تغطيه المنطقة لكل منطقة مدرجة. كلما ارتفعت النسبة المئوية للقواعد المطابقة ، زاد الحفظ للمنطقة ذات الاهتمام. - اتبع نفس الاستراتيجيات الملاحية المفصلة في القسم 4 لتحليل التسلسل.
6. توليد محاذاة تسلسل متعددة الأنواع للبروتينات الدقيقة ذات الأهمية
- انقر على الجين الذي يهم في مسار GENCODE على متصفح UCSC Genome Browser (المشار إليه في الشكل 1A بمربع أزرق) للانتقال إلى صفحة وصف الجين.
- تحت عنوان التسلسل والروابط إلى الأدوات وقواعد البيانات ، انقر فوق الرابط الموجود في الجدول الذي يقرأ FASTA الأنواع الأخرى.
- انقر على المربعات المرتبطة بالأنواع ذات الأهمية لتحديدها. انقر على إرسال. انسخ التسلسلات التي تظهر في أسفل الصفحة بتنسيق FASTA والصقها في مستند معالجة نصوص.
- افتح نافذة متصفح ثانية وانتقل إلى أداة Clustal Omega Multi Sequence Alignment 52 على موقع المعهد الأوروبي للمعلوماتية الحيوية (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- الصق ملفات التسلسل التي لا تزال موجودة في الحافظة في المربع الموجود في الخطوة 1 التي تقرأ التسلسلات بأي تنسيق معتمد. مرر إلى أسفل الصفحة وانقر على إرسال. انظر أسفل النتائج المحاذاة (بالخط الأسود) للرموز التي تشير إلى درجة حفظ كل حمض أميني (يتم تعريف الرموز في الجدول 1).
ملاحظة: قد يستغرق إنشاء المحاذاة عدة دقائق. - لعرض خصائص الأحماض الأمينية في اللون ، انقر فوق رابط إظهار الألوان مباشرة فوق التسلسلات لتلوين الأحماض الأمينية وفقا لخصائصها (المحددة في الجدول 2).
- انسخ محاذاة التسلسل والصقها في برنامج معالجة النصوص أو عرض الشرائح لإنشاء ملف شكل أو رسم توضيحي (على سبيل المثال، الشكل 2).
ملاحظة: استخدم خطا أحادي المسافة للمحاذاة مثل Courier. - لعرض المخرجات الأخرى من صفحة نتائج Clustal Omega ، انقر فوق علامات التبويب المناسبة (أي شجرة الدليل أو شجرة التكاثر).
- انقر فوق علامة التبويب "مشاهدو النتائج " للحصول على خيارات لعرض معلومات التسلسل باستخدام Jalview ، وهو برنامج مجاني متخصص في تحرير محاذاة التسلسل المتعدد والتصور والتحليل55 ، أو للوصول إلى روابط مباشرة إلى MView و Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
هنا سنستخدم ميتوريغولين البروتين الدقيق الذي تم التحقق منه (Mtln) كمثال لتوضيح كيف سيولد sORF المحفوظ درجة PhyloCSF إيجابية يمكن تصورها وتحليلها بسهولة على متصفح UCSC Genome. تم شرح الميتوريجولين سابقا على أنه حمض نووي ريبي غير مشفر (معرف الجين البشري سابقا LINC00116 ومعرف جين الفأر 1500011K16Rik). لعبت طرق تحليل الجينوم المقارن وتحليل التسلسل دورا حاسما في اكتشافها الأولي40،57،58،59،60،61 ، مما يسلط الضوء على قوة هذه الأساليب. على سبيل المثال، سيتم استخدام تجميع الماوس GRCm38/mm10 (ديسمبر 2011). يمكن إجراء البحث باستخدام معرفات الجينات (الميتوريغولين ، Mtln) أو موضع الجين (chr2: 127،791،364-127،792،496) كما هو موضح في قسم البروتوكول 2. بدلا من ذلك ، يمكن البحث في تسلسل الأحماض الأمينية للميتوريغولين (كما هو موضح في الشكل 2) باستخدام أداة BLAT (الموضحة في قسم البروتوكول 3).
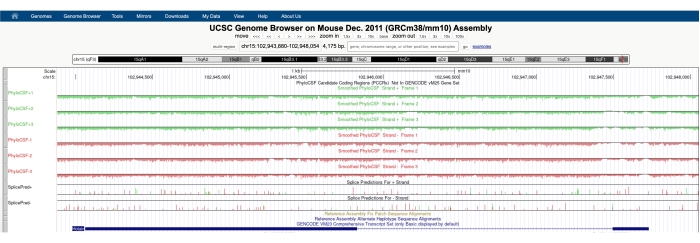
ستظهر شاشة مشابهة لتلك الموضحة في الشكل 1A مع ظهور PhyloCSF Track Hub في الجزء العلوي من الشاشة. يتم تصوير مسارات PhyloCSF الملساء (الملساء بنموذج ماركوف المخفي الذي يحدد احتمال ترميز كل كودون) على أنها ستة مسارات إجمالية ، مع ثلاثة مسارات تتوافق مع الشريط الزائد للحمض النووي (مصور باللون الأخضر ك PhyloCSF +1 و +2 و +3) وثلاثة مسارات تتوافق مع الشريط الناقص من الحمض النووي (مصور باللون الأحمر باسم PhyloCSF -1 ، -2 و-3). تمثل هذه المسارات إطارات القراءة الثلاثة المحتملة للجين محل الاهتمام في كل اتجاه. في نافذة المتصفح ، يتم تصوير exons على أنها مستطيلات زرقاء متصلة بخطوط أفقية زرقاء رقيقة ، والتي تمثل الإنترونات. تشير رؤوس الأسهم الموجودة على المناطق الإنترونية إلى الاتجاه الذي يتم نسخ الجين فيه (وبالتالي ، أي خيط يجب التركيز عليه للحصول على درجة PhyloCSF). على سبيل المثال Mtln في الشكل 1 ، تشير رؤوس الأسهم الإنترونية إلى اليسار. لذلك ، يتم نسخ جين Mtln من الشريط الناقص للحمض النووي ، ويتم تصوير درجة PhyloCSF ذات الصلة في المسارات -1 و -2 و -3 (باللون الأحمر).
يتم تصوير كل مسار PhyloCSF على أنه خط أسود رفيع مع مناطق تسجيل سلبية مصورة باللون الأخضر الفاتح / الأحمر أسفل الخط ومناطق تسجيل إيجابية مشار إليها باللون الأخضر الداكن / الأحمر فوق الخط. كما هو موضح في المقدمة ، تشير درجة PhyloCSF الإيجابية إلى منطقة محفوظة من المحتمل أن تكون مشفرة. لاحظ أنه بالنسبة لمناطق ترميز البروتين ذات الحفاظ على تسلسل عال بشكل خاص ، فإنها غالبا ما تسجل أيضا نتائج إيجابية على الشريط المضاد للإحساس ؛ ومع ذلك ، فإن درجة PhyloCSF عادة ما تكون أعلى على الخيط الصحيح. على سبيل المثال ، يمكن ملاحظة ذلك في الشكل 1 ل Mtln حيث يسجل تسلسل الترميز الصحيح درجات عالية جدا في مسار PhyloCSF -1 ، كما يولد الشريط المضاد للإحساس (مسار PhyloCSF +2) درجة إيجابية. كما هو موضح في الشكل 1A (المشار إليه بالصندوق الأسود) ، هناك منطقة في الإكسون الأول من Mtln تسجل درجات عالية جدا على مسار PhyloCSF -1 ، مما يشير إلى أن هذا قد يتوافق مع منطقة ترميز. لدراسة هذه المنطقة بمزيد من التفصيل ، من المفيد تكبير المنطقة وتكبيرها (الشكل 1B). كما هو موضح في الشكل 1C ، D ، تبدأ منطقة التسجيل الإيجابية في الإكسون الأول من Mtln مباشرة فوق كودون البدء (الشكل 1C) وتنتهي عند كودون التوقف (الشكل 1D) ، مما يشير إلى أن ORF هذا محفوظ بشكل كبير ويشير بقوة إلى أنه ترميز ORF. نظرا لأن Mtln موجود على الشريط الناقص من الحمض النووي ، يتم عرض كودونات البدء والتوقف كمكمل عكسي للكودون (أي ، يظهر كودون بدء ATG على أنه CAT [الشكل 1C] ويظهر كودون إيقاف TGA على أنه TCA [الشكل 1D]).
بالإضافة إلى استخدام PhyloCSF للبحث عن المناطق المحفوظة ذات إمكانات ترميز البروتين الدقيق ، يمكن أيضا تطبيق هذه التقنية كتحليل أول للحمض النووي الريبي غير المشفر المفترض لاستبعاد وجود ORF محفوظ ، وبالتالي توفير الدعم لتعليق توضيحي غير مشفر. على سبيل المثال، يظهر تحليل lncRNA HOTAIR62,63 المميز جيدا باستخدام PhyloCSF درجة سلبية في جميع أنحاء الجين بأكمله عبر جميع المسارات الستة (الشكل 3)، مما يشير بقوة إلى عدم الحفاظ على التسلسل ويوفر الدعم بأن HOTAIR مشروح بشكل صحيح على أنه حمض نووي ريبي غير مشفر.
كما هو موضح بوضوح في الشكل 1 ، يقع الترميز الكامل ORF للميتوريغولين داخل إكسون واحد ، مما ينتج عنه قراءة بسيطة ومباشرة من قبل PhyloCSF مع منطقة تسجيل واحدة ، دون انقطاع ، بشكل إيجابي. ومع ذلك ، فإن بيانات محور المسار PhyloCSF ليست دائما واضحة وسهلة التفسير. على سبيل المثال ، يصور البروتين الدقيق mitolamban / Stmp1 / Mm47 المشفر بواسطة جين الماوس 1810058I24Rik 47,64,65 ORF محفوظا يمتد عبر ثلاثة إكسونات (الشكل 4A) ، وتقفز درجة PhyloCSF الإيجابية من المسار +2 في exon 1 (الشكل 4B) إلى المسار +3 في exon 2 (الشكل 4C) ، ثم العودة إلى المسار +2 في exon 3 (الشكل 4D) ). في حين أن هذا يبدو مربكا للوهلة الأولى ، إلا أن التفسير واضح تماما. يسجل PhyloCSF إطارات القراءة الستة المحتملة (ثلاثة على الشريط الزائد للحمض النووي وثلاثة على الشريط الناقص) للمناطق الجينومية دون النظر في بنية exon / intron المحددة لكل جين. لذلك ، فإنه يحتفظ بمعلومات التسلسل الإنترونيكي في دورية 3-nucleotide لإطارات القراءة. وبالتالي ، إذا كان الإنترون يحتوي على عدد من النيوكليوتيدات غير القابلة للقسمة على ثلاثة (أي ثلاثة نيوكليوتيدات / كودون) ، فإن إطار قراءة PhyloCSF سوف يقفز من مسار إلى آخر.
أخيرا ، يمكن أيضا استخدام PhyloCSF بشكل فعال لتحديد العديد من ORFs المشفرة المميزة داخل جزيء RNA واحد. على سبيل المثال ، يتم ترميز البروتين الدقيق MIEF1 (MIEF1-MP) داخل UTR 5 'من عامل استطالة الميتوكوندريا 1 (MIEF1)66 (الشكل 5). عندما يتم تحليل المنطقة الجينومية MIEF1 بواسطة PhyloCSF ، يمكن بسهولة ملاحظة درجة PhyloCSF إيجابية منفصلة تتوافق مع MIEF1-MP (الشكل 5C) في المنبع من CDS الرئيسية ل MIEF1 (الشكل 5B). يتم توفير مزيد من المناقشة حول MIEF1 والبروتين الدقيق المرتبط به (MIEF1-MP) أدناه في المناقشة إلى جانب ملخص لنقاط القوة والضعف في الأساليب والبروتوكولات الموضحة في هذه المقالة.

الشكل 1: يشير تحليل PhyloCSF لجين الميتوريغولين (Mtln) إلى منطقة حفظ عالية التسلسل تتوافق مع بروتين دقيق تم التحقق منه. (أ) تظهر لقطات شاشة لمتصفح UCSC Genome Browser و PhyloCSF Tracks أن Mtln يحتوي على إكسون اثنين وإنترون واحد. تشير رؤوس الأسهم داخل الإنترون إلى اليسار ، مما يشير إلى أن جين Mtln يتم نسخه من الشريط الناقص للحمض النووي ، وبالتالي يتم عرض درجات PhyloCSF ذات الصلة في المسارات -1 و -2 و -3 (باللون الأحمر). يتم تضمين تسلسل ترميز الميتوريغولين الكامل داخل Exon 1 ويسجل درجات عالية على مسار PhyloCSF -1 (B). يمكن ملاحظة كودون البدء المحفوظ بوضوح في بداية منطقة التسجيل الإيجابية في مسار PhyloCSF -1 (C) ، والذي يتم تمييزه بمربع أخضر (CAT ، مكمل عكسي ATG). بالإضافة إلى ذلك ، يشار إلى كودون التوقف المحفوظ (TCA ، TGA المكمل العكسي) بمربع أحمر في اللوحة (D) ، والذي يتماشى مع نهاية منطقة PhyloCSF ذات التسجيل الإيجابي. يمكن العثور على معلومات مفصلة حول جين Mtln بالنقر فوق معرف جين Mtln داخل المربع الأزرق (كما هو موضح في اللوحة A). تجدر الإشارة إلى أن مناطق ترميز البروتين المحفوظة للغاية غالبا ما تسجل أيضا نتائج إيجابية على الشريط المضاد للإحساس (كما هو موضح هنا في مسار PhyloCSF +2 ل Mtln). ومع ذلك ، فإن درجة PhyloCSF عادة ما تكون أعلى على الشريط الصحيح (مسار PhyloCSF -1 في هذا المثال). يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.

الشكل 2: محاذاة تسلسل الأنواع المتعددة للميتوريغولين البروتين الدقيق المتولد باستخدام برنامج كلوستال أوميغا. تم استخراج تسلسلات الأحماض الأمينية للميتوريغولين للأنواع الثمانية المشار إليها على النحو المفصل في قسم البروتوكول 6 ومحاذاة مع أداة محاذاة التسلسل المتعدد Clustal Omega. يشار إلى خصائص الأحماض الأمينية حسب اللون (الأحمر ، الصغير / الكارهة للماء ؛ الأزرق ، الحمضي ؛ الأرجواني ، الأساسي ؛ الأخضر ، الهيدروكسل / السلفهيدرول / الأمين) (يتم تعريفه بشكل أكبر في الجدول 2). تشير الرموز الموجودة أسفل الأحماض الأمينية إلى درجة الحفظ (العلامات النجمية ، المخلفات المحفوظة بالكامل ؛ القولون ، الأحماض الأمينية ذات الخصائص المتشابهة بقوة ؛ الفترات ، الحفظ بين مجموعات ذات الخصائص المتشابهة بشكل ضعيف) (مفصلة على نطاق واسع في الجدول 1). يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
الشكل 3: لقطة شاشة لمسارات PhyloCSF ل RNA Hotair الطويل غير المشفر الذي تم التحقق منه يظهر عدم وجود حفظ تسلسل في جميع أنحاء موقعه الجينومي. تشير رؤوس الأسهم في المنطقة الإنترونية من Hotair إلى اليسار ، مما يشير إلى أن lncRNA يتم نسخه من الشريط السلبي للحمض النووي ، وبالتالي يجب أن تكون مسارات PhyloCSF -1 و -2 و -3 محور التحليل. لاحظ أن درجة PhyloCSF سلبية في جميع أنحاء الجين بأكمله (لجميع المسارات الستة) ، مما يشير إلى عدم الحفاظ على التسلسل ، مما يدعم التعليق التوضيحي المناسب كحمض نووي غير مشفر. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.

الشكل 4: تحليل PhyloCSF لجين الفأر 1810058I24Rik ، الذي يشفر البروتين الدقيق mitolamban / Stmp1 / Mm47. (A) يتكون جين الماوس 1810058I24Rik من ثلاثة إكسونات ، وتشير رؤوس الأسهم في المناطق الإنترونية إلى اليمين ، مما يشير إلى أنه يتم نسخه على الشريط الزائد من الحمض النووي ، وبالتالي يجب تحليل مسارات PhyloCSF +1 و +2 و +3. يمتد تسلسل ترميز البروتين الدقيق المحفوظ على جميع الإكسونات الثلاثة، بدءا من إكسون 1 (ب)، وقراءة من خلال إكسون 2 (ج)، وانتهاء بإكسون 3 (د). لاحظ أن نتيجة PhyloCSF الإيجابية موجودة على المسار +2 في exon 1 ، والمسار +3 في exon 2 ، والمسار +2 في exon 1. سبب حركة النتيجة الإيجابية من مسار إلى آخر هو أن PhyloCSF يحلل إطارات القراءة الستة المحتملة لتسلسل الحمض النووي بشكل مستقل عن بنية الإكسون / الإنترون للجين. لذلك ، فإن الإنترون الذي يحتوي على عدد من النيوكليوتيدات غير القابلة للقسمة على ثلاثة (ثلاثة نيوكليوتيدات / كودون) سيؤدي إلى تحول في إطار القراءة إلى مسار مختلف. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.

الشكل 5: تحليل الموضع الجينومي Mief1 باستخدام PhyloCSF يحدد منطقة ذات إمكانات ترميز البروتين في UTR 5 'التي هي مستقلة عن CDS Mief1 الرئيسي على الحمض النووي الريبي المشترك. وقد تبين أن هذا المنبع المحفوظ ORF (uORF) يشفر بروتينا دقيقا يسمى Mief1-MP. (أ) نظرة عامة على الموقع الجينومي Mief1. تشير رؤوس الأسهم في الإنترونات إلى اليمين ، مما يشير إلى أن Mief1 يتم نسخه من الشريط الزائد للحمض النووي (ركز على مسارات PhyloCSF +1 و +2 و +3 لتحديد إمكانات الترميز). يشفر Mief1 CDS الرئيسي بروتين الأحماض الأمينية 463 ويظهر في اللوحة (B). ومع ذلك ، هناك أيضا ORF متميز محفوظ في المنبع داخل UTR 5 'من Mief1 الذي يشفر بروتين دقيق فريد من نوعه من الأحماض الأمينية 70 يسمى Mief1-MP (C). كما رأينا في اللوحة C ، فإن Mief1-MP لديه كودون بدء وإيقاف محفوظ خاص به داخل Mief1 5 'UTR ، ويسجل ORF درجات عالية جدا على مسار PhyloCSF +1 ، مما يوفر دليلا قويا على أنه يشفر البروتين الدقيق الوظيفي. الاختصارات: ORF = إطار قراءة مفتوح. uORF = المنبع ORF ؛ UTR = منطقة غير مترجمة; CDS = تسلسل الترميز. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
| رمز | مستوى الحفاظ على الأحماض الأمينية | الأحماض الأمينية المجمعة |
| العلامة النجمية (*) | بقايا محفوظة بالكامل | غير قابل للتطبيق (بقايا مفردة محفوظة بالكامل) |
| القولون (:) | المجموعات ذات الخصائص المتشابهة بشدة | ستا. NEQK; NHQK; NDEQ; QHRK; ميلف. جبهة مورو الإسلامية للتحرير; هاي. FYW |
| الفترة (.) | المجموعات ذات الخصائص المتشابهة بشكل ضعيف | وكالة الفضاء الكندية; مركبة الدفع الرباعي. SAG; STNK. STPA; SGND; SNDEQK; NDEQHK; نيقهرك. FVLIM. HFY |
| المسافة (بدون رمز) | لا تشابه | غير قابل للتطبيق (لا يوجد تشابه) |
الجدول 1: تعريفات رموز الإجماع لمحاذاة التسلسل المتعدد الناتجة عن Clustal Omega. تم إنشاء محاذاة تسلسل الأنواع المتعددة الموضحة في الشكل 2 باستخدام Clustal Omega52. الاختصارات: سيرين (S) ، ثريونين (T) ، ألانين (A) ، أسباراجين (N) ، حمض الجلوتاميك (E) ، الجلوتامين (Q) ، ليسين (K) ، حمض الأسبارتيك (D) ، أرجينين (R) ، ميثيونين (M) ، آيسولوسين (I) ، ليوسين (L) ، فينيل ألانين (F) ، هيستيدين (H) ، التيروزين (Y) ، التربتوفان (W) ، السيستين (C) ، فالين (V) ، الجلايسين (G) ، البرولين (P).
| لون الخط | مال | بقايا الأحماض الأمينية [اختصار] |
| أحمر | صغير، مسعور | ألانين [A]، فالين [V]، فينيل ألانين [F]، برولين [P]، ميثيونين [M]، إيزولوسين [I]، ليوسين [L]، تريبتوفان [W] |
| أزرق | الحمضيه | حمض الأسبارتيك [D]، حمض الجلوتاميك [E] |
| الارجواني | أساسي | أرجينين [R]، ليسين [K] |
| أخضر | هيدروكسل، سلفهيدريل، أمين، +G | سيرين [S]، ثريونين [T]، التيروزين [Y]، الهيستيدين [H]، السيستين [C]، الأسباراجين [N]، الجلايسين [G]، الجلوتامين [Q] |
الجدول 2: خصائص الأحماض الأمينية المبينة في الشكل 2. تم استخدام Clustal Omega52 لتوليد محاذاة التسلسل المتعدد الموضحة في الشكل 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
يوفر البروتوكول المعروض هنا تعليمات مفصلة حول كيفية استجواب المناطق الجينومية ذات الأهمية لإمكانات ترميز البروتين الدقيق باستخدام PhyloCSF على متصفح UCSC Genome Browser 48,49,50,51 سهل الاستخدام. كما هو مفصل أعلاه ، PhyloCSF هي خوارزمية جينوم مقارنة قوية تدمج النماذج الجينية وترددات استبدال الكودون لتحديد التوقيعات التطورية النموذجية لجينات ترميز البروتين48,49. تم استخدام PhyloCSF على نطاق واسع لتحديد البروتينات الدقيقة الوظيفية في المناطق الجينومية التي سبق شرحها على أنها غير مشفرة 38,39,40,41,42,43,44,45,46,47 ، وقد ثبت أن هذا النهج يتفوق على طرق الجينوم المقارنة الأخرى للتسلسلات القصيرة مثل البروتينات الدقيقة الصغيرة مثل 13 حمضا أمينيا وللإكسونات الصغيرة من البروتينات الأساسية35،48،49. ومن الجدير بالذكر أن فائدة PhyloCSF كطريقة قوية لتحديد تسلسلات ترميز البروتين الوظيفية عن طريق الحفظ التطوري تمتد إلى ما هو أبعد من الأنواع الفقارية واللافقاريات ، وقد تم تطبيقها مؤخرا على الجينوم الفيروسي لاستجواب قدرة ترميز البروتين بنجاح في جينوم SARS-CoV-267.
بالإضافة إلى تحديد تسلسلات الترميز المفترضة داخل الحمض النووي الريبي غير المشفر المشروح ، فإن ميزة PhyloCSF هي أنه يمكنه أيضا الكشف بشكل موثوق عن البروتينات الدقيقة المحفوظة المشفرة بواسطة ORFs داخل المناطق غير المترجمة المشروحة (UTRs) لجينات ترميز البروتين الأساسية ، بما في ذلك كل من 5 'المنبع و 3' ORFs المصب (uORFs و dORFs ، على التوالي)8,19,66,68 . على سبيل المثال ، يتم ترميز البروتين الدقيق MIEF1 (MIEF1-MP) في UTR 5 'من عامل استطالة الميتوكوندريا 1 (MIEF1)66. في حالة MIEF1-MP ، لوحظت درجة PhyloCSF إيجابية منفصلة تتوافق مع MIEF1-MP في المنبع من ORF التي تشفر MIEF1 (الشكل 5). في حين أن بعض البروتينات الدقيقة المشفرة uORF تتفاعل مباشرة مع البروتينات الأساسية في المصب على الحمض النووي الريبوزي المرسال المشترك ، (على سبيل المثال MIEF1-MP و MIEF1) ، يعمل البعض الآخر بشكل مستقل عن البروتين المشفر بواسطة CDS الرئيسي66,68. لذلك ، عند توصيف البروتينات الدقيقة المشفرة ب uORF ، لا ينبغي افتراض أنها تعمل عن طريق التنظيم المباشر لمنتج البروتين النهائي.
في حين أن PhyloCSF لديه العديد من نقاط القوة الواضحة كأداة لتحديد تسلسلات ترميز البروتين الدقيق المحفوظة ، فمن المهم التعرف على العديد من القيود على هذه الطريقة. أولا، في حين أن حفظ التسلسل يشير بقوة إلى أن منطقة الجينوم قد خضعت للاختيار الوظيفي وبالتالي فهي ترميز، فإن عدم وجود حفظ قوي وما ينتج عن ذلك من درجة PhyloCSF سلبية لا يستبعد بشكل قاطع إمكانات الترميز لتسلسل معين. وبعبارة أخرى، فإن الاعتماد حصرا على PhyloCSF قد يؤدي إلى الإشراف على ORFs المترجمة التي لا يتم حفظها بقوة ولكنها لا تزال تنتج بروتينات دقيقة وظيفية. ومن الجدير بالذكر أن المناطق الجينومية ذات درجات الحفظ المنخفضة أو الحفظ السلبية يمكن أن تتوافق مع مناطق الترميز الخاصة بالأنواع أو تلك الخاصة بالجينات "الشابة" التطورية عن طريق اختلاف التسلسل أو ولادة الجينات الجديدة 46,69,70,71,72,73,74. على سبيل المثال ، لا يتم تسجيل البروتين الدقيق في أسرع وقت ممكن ، والذي يتم ترميزه بواسطة ما كان يعتقد سابقا أنه الحمض النووي الريبي البشري غير المشفر LINC00467 ، بشكل إيجابي بواسطة PhyloCSF لأن تسلسل الأحماض الأمينية محفوظ فقط في الثدييات الأعلى75. بالإضافة إلى ذلك ، حددت الدراسات الحديثة العديد من البروتينات الدقيقة الخاصة بالإنسان ، بما في ذلك واحدة مشفرة بواسطة lncRNA RP3-527G5.1 بين الجينات ، والتي لا تولد درجة PhyloCSF إيجابية 68,72. في هذا الصدد ، لا يمكن تفسير عدم وجود درجة PhyloCSF إيجابية على أنها دليل على وجود منطقة غير مشفرة ويجب تفسيرها بحذر.
الاعتبار الثاني الذي يجب مراعاته عند استخدام PhyloCSF هو أنه على الرغم من أن النتيجة الإيجابية توحي بشكل كبير بالاختيار الوظيفي والقدرة على ترميز البروتين ، إلا أن هذا الخط من الأدلة لا يمكن أن يقف بمفرده ويجب التحقق من صحته تجريبيا. ومن الأمثلة على الطرق التي يمكن استخدامها لتوليد أدلة داعمة للتعبير المستقر عن البروتين الدقيق الكشف عن البروتين المفترض عن طريق قياس الطيف الكتلي أو النشاف الغربي باستخدام جسم مضاد يثار ضد تسلسل البروتين الدقيق محل الاهتمام. بدلا من ذلك ، نظرا لأنه قد يكون من الصعب توليد أجسام مضادة موثوقة للبروتينات الدقيقة بسبب عدم وجود خيارات تسلسل للمستضد الأمثل ، فمن الممكن أيضا استخدام CRISPR / Cas9 ومسار الإصلاح الموجه بالتماثل (HDR) لإدخال علامة epitope في الموضع الداخلي في الإطار مع تسلسل البروتين الدقيق المفترض ، وبالتالي تسهيل الكشف عن البروتين محل الاهتمام باستخدام جسم مضاد عالي التقارب (على سبيل المثال ، العلم ، ها ، V5 ، Myc) 18. القيد الأخير ل PhyloCSF هو أنه على الرغم من أنه مدمج حاليا في العديد من التجمعات الجينومية الشائعة الاستخدام ، بما في ذلك الإنسان العاقل (الإنسان hg19 ، hg38) ، العضلات العضلية (الفأر mm10 ، mm39) ، Gallus gallus (الدجاج ، galGal4 ، galGal6) ، ذبابة الفاكهة melanogaster (ذبابة الفاكهة ، dm6) ، Caenorhabditis elegans (الديدان الخيطية ، ce11) ، و SARS-CoV-2 (wuhCor1) ، لا يزال هناك العديد من الأنواع التي لا يمكن الاستعلام عنها حاليا مباشرة على متصفح الجينوم UCSC.
يمكن أن يساعد تحديد المجالات المحفوظة أو خصائص التسلسل داخل البروتينات الدقيقة المحددة في زيادة الثقة في أهميتها الوظيفية وتوفير بعض البصيرة في وظيفتها المفترضة. هنا نقدم توصيات لأدوات وموارد محددة يمكن استخدامها لتحليل تسلسلات الأحماض الأمينية للبروتين الدقيق المحددة بمزيد من التفصيل للحصول على مثل هذه البصيرة. الأدوات المحددة المدرجة أدناه (والملخصة في جدول المواد) متاحة مجانا للجمهور ، وقد وجدنا أنها سهلة الاستخدام وقوية بشكل خاص في دراسات البروتين الدقيق18،38،39،40،41،47. بالإضافة إلى الأدوات الموضحة هنا ، هناك العديد من الموارد الإضافية التي يمكن العثور عليها في بوابات موارد المعلوماتية الحيوية مثل Expasy (https://www.expasy.org) و EMBL-EBI (https://www.ebi.ac.uk/services/all). ومع ذلك ، فإن تفصيل تفاصيل كل أداة من الأدوات داخل هذه المستودعات خارج نطاق هذه المقالة. هنا نوصي بالموارد التالية.
أولا ، يحلل TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) تسلسلات البروتين ذات الأهمية لوجود مجالات عبر الغشاء. ومن الجدير بالذكر أن عددا من البروتينات الدقيقة التي تم تمييزها وظيفيا حتى الآن تحتوي على مجالات عبر غشاء أحادية المرور ، مما يسهل توطينها في مناطق الغشاء ويتيح تنظيمها المباشر للقنوات الأيونية والمبادلات والإنزيمات المرتبطة بالغشاء30. ثانيا، يعد بحث المجال المحفوظ77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) الصادر عن المركز الوطني لمعلومات التكنولوجيا الحيوية (NCBI) أداة شائعة تستخدم لتحديد المجالات المحفوظة داخل تسلسلات النيوكليوتيدات البروتينية أو المشفرة. ثالثا ، توفر قاعدة بيانات عائلة البروتين (Pfam)78 (http://pfam.xfam.org) محاذاة وتصنيفات لعائلات البروتين ومجالاته. رابعا ، WoLF PSORT79 (https://wolfpsort.hgc.jp/) هي أداة يمكن استخدامها للتنبؤ بتوطين البروتين تحت الخلوي. خامسا ، COXPRESdB80 هي قاعدة بيانات للتعبير الجيني المشترك (https://coxpresdb.jp) توفر علاقات جينية منظمة بشكل مشترك لتقدير وظائف الجينات. أخيرا ، SignalP 6.081 هو برنامج تنبؤ يستخدم على نطاق واسع (https://services.healthtech.dtu.dk/service.php?SignalP) يتعرف على وجود تسلسل ببتيد الإشارة ويتنبأ بموقع موقع الانقسام.
باختصار ، يمكن استخدام الطرق الموضحة هنا لتحليل المناطق الجينومية ذات الأهمية بشكل فعال لإمكانات ترميز البروتين باستخدام PhyloCSF على متصفح UCSC Genome. هذه الأساليب يمكن الوصول إليها بسهولة ويمكن تعلمها بسهولة وتطبيقها بكفاءة من قبل الأفراد دون تدريب مسبق أو خبرة في المعلوماتية الحيوية أو علم الجينوم المقارن. كما هو موضح هنا بالتفصيل ، فإن PhyloCSF هي أداة قوية يمكن تطبيقها كتحليل للمرور الأول للمساعدة في التمييز بين ترميز البروتين مقابل الجينات غير المشفرة في الفقاريات واللافقاريات والجينوم الفيروسي ، ونقاط القوة في هذا النهج تفوق بشكل كبير نقاط الضعف الملحوظة.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
ويعلن صاحبا البلاغ أنه ليس لديهما مصالح مالية منافسة.
Acknowledgments
تم دعم هذا العمل من خلال منح من المعاهد الوطنية للصحة (HL-141630 و HL-160569) ومؤسسة سينسيناتي لأبحاث الأطفال (جائزة الوصي).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
