

Summary
यहां वर्णित प्रोटोकॉल उपयोगकर्ता के अनुकूल यूसीएससी जीनोम ब्राउज़र पर फिलोस्सफ का उपयोग करके माइक्रोप्रोटीन-कोडिंग क्षमता के लिए ब्याज के जीनोमिक क्षेत्रों का विश्लेषण करने के तरीके पर विस्तृत निर्देश प्रदान करता है। इसके अतिरिक्त, पहचाने गए माइक्रोप्रोटीन की अनुक्रम विशेषताओं की जांच करने के लिए कई उपकरणों और संसाधनों की सिफारिश की जाती है ताकि उनके पुटेटिव कार्यों में अंतर्दृष्टि प्राप्त की जा सके।
Abstract
अगली पीढ़ी के अनुक्रमण (एनजीएस) ने जीनोमिक्स के क्षेत्र को आगे बढ़ाया है और कई जानवरों की प्रजातियों और मॉडल जीवों के लिए पूरे जीनोम अनुक्रमों का उत्पादन किया है। हालांकि, अनुक्रम जानकारी के इस धन के बावजूद, व्यापक जीन एनोटेशन प्रयास चुनौतीपूर्ण साबित हुए हैं, खासकर छोटे प्रोटीन के लिए। विशेष रूप से, पारंपरिक प्रोटीन एनोटेशन विधियों को जानबूझकर जीनोम में नकली नॉनकोडिंग एसओआरएफ की तेजी से अधिक संख्या को फ़िल्टर करने के लिए लंबाई में 300 न्यूक्लियोटाइड से कम शॉर्ट ओपन रीडिंग फ्रेम (एसओआरएफ) द्वारा एन्कोडेड प्रोटीन को बाहर करने के लिए डिज़ाइन किया गया था। नतीजतन, माइक्रोप्रोटीन (लंबाई में <100 अमीनो एसिड) नामक सैकड़ों कार्यात्मक छोटे प्रोटीन को गलत तरीके से नॉनकोडिंग आरएनए के रूप में वर्गीकृत किया गया है या पूरी तरह से अनदेखा किया गया है।
यहां हम विकासवादी संरक्षण के आधार पर माइक्रोप्रोटीन-कोडिंग क्षमता के लिए जीनोमिक क्षेत्रों से पूछताछ करने के लिए मुफ्त, सार्वजनिक रूप से उपलब्ध जैव सूचनात्मक उपकरणों का लाभ उठाने के लिए एक विस्तृत प्रोटोकॉल प्रदान करते हैं। विशेष रूप से, हम उपयोगकर्ता के अनुकूल कैलिफोर्निया सांताक्रूज विश्वविद्यालय (यूसीएससी) जीनोम ब्राउज़र पर फाइटोलैनेटिक कोडन प्रतिस्थापन आवृत्तियों (फिलोस्सफ) का उपयोग करके अनुक्रम संरक्षण और कोडिंग क्षमता की जांच करने के तरीके पर चरण-दर-चरण निर्देश प्रदान करते हैं। इसके अतिरिक्त, हम अमीनो एसिड अनुक्रम संरक्षण की कल्पना करने के लिए पहचाने गए माइक्रोप्रोटीन अनुक्रमों के कई प्रजातियों के संरेखण को कुशलतापूर्वक उत्पन्न करने के लिए चरणों का विस्तार करते हैं और अनुमानित डोमेन संरचनाओं सहित माइक्रोप्रोटीन विशेषताओं का विश्लेषण करने के लिए संसाधनों की सिफारिश करते हैं। इन शक्तिशाली उपकरणों का उपयोग नॉनकैनोनिकल जीनोमिक क्षेत्रों में पुटेटिव माइक्रोप्रोटीन-कोडिंग अनुक्रमों की पहचान करने में मदद करने के लिए किया जा सकता है या ब्याज के नॉनकोडिंग ट्रांसक्रिप्ट में ट्रांसलेशनल क्षमता के साथ एक संरक्षित कोडिंग अनुक्रम की उपस्थिति को खारिज करने के लिए किया जा सकता है।
Introduction
जीनोम में कोडिंग तत्वों के पूर्ण सेट की पहचान मानव जीनोम परियोजना की दीक्षा के बाद से एक प्रमुख लक्ष्य रहा है, और जैविक प्रणालियों की समझ और आनुवंशिक-आधारित बीमारियोंके एटियलजि की दिशा में एक केंद्रीय उद्देश्य बना हुआ है 1,2,3,4। एनजीएस तकनीकों में प्रगति ने कशेरुक, अकशेरुकी, खमीर और पौधों सहित जीवों की एक विस्तृत संख्या के लिए पूरे जीनोम अनुक्रमों का उत्पादन कियाहै 5. इसके अतिरिक्त, उच्च-थ्रूपुट ट्रांसक्रिप्शनल अनुक्रमण विधियों ने सेलुलर ट्रांसक्रिप्टोम की जटिलता को और प्रकट किया है, और प्रोटीन-कोडिंग और नॉनकोडिंग फ़ंक्शंस 6,7 दोनों के साथ हजारों उपन्यास आरएनए अणुओं की पहचान की है। अनुक्रम जानकारी की इस विशाल मात्रा को डिकोड करना एक सतत प्रक्रिया है, और चुनौतियां व्यापक जीन एनोटेशनप्रयासों के साथ बनी हुई हैं 8.
राइबोसोम प्रोफाइलिंग 9,10 और पॉली-राइबोसोमअनुक्रमण 11 सहित ट्रांसलेशनल प्रोफाइलिंग विधियों के हालिया विकास ने सबूत प्रदान किए हैं कि सैकड़ों गैर-विहित अनुवाद घटनाएं पूरे जीनोम में वर्तमान में अप्रकाशित एसओआरएफ के लिए मैप करती हैं, जिसमें माइक्रोप्रोटीन या माइक्रोपेप्टाइड्स12,13,14,15,16 नामक छोटे प्रोटीन उत्पन्न करने की क्षमता होती है। 17. माइक्रोप्रोटीन बहुमुखी प्रोटीन के एक उपन्यास वर्ग के रूप में उभरे हैं, जिन्हें पहले उनके छोटे आकार (<100 अमीनो एसिड) और शास्त्रीय प्रोटीन-कोडिंग जीन विशेषताओं 8,12,18,19,20 की कमी के कारण मानक जीन एनोटेशन विधियों द्वारा अनदेखा किया गया था। खमीर21,22, मक्खियों 17,23,24, और स्तनधारियों 25,26,27,28 सहित लगभग सभी जीवों में माइक्रोप्रोटीन का वर्णन किया गया है, और विकास, चयापचय और तनाव सिग्नलिंग सहित विभिन्न प्रक्रियाओं में महत्वपूर्ण भूमिका निभाने के लिए दिखाया गया है 19,20,29, 30,31,32,33,34. इस प्रकार, कार्यात्मक छोटे प्रोटीन के इस लंबे समय से अनदेखी वर्ग के अतिरिक्त सदस्यों के लिए जीनोम को खदान जारी रखना अनिवार्य है।
माइक्रोप्रोटीन के जैविक महत्व की व्यापक मान्यता के बावजूद, जीन का यह वर्ग जीनोम एनोटेशन में काफी कम प्रतिनिधित्व करता है, और उनकी सटीक पहचान एक सतत चुनौती बनी हुई है जिसने क्षेत्र में प्रगति में बाधा डाली है। माइक्रोप्रोटीन-कोडिंग अनुक्रमों की पहचान करने से जुड़ी कठिनाइयों को दूर करने के लिए हाल ही में विभिन्न कम्प्यूटेशनल टूल और प्रयोगात्मक विधियों को विकसित किया गया है (कई व्यापक समीक्षाओं 8,35,36,37 में बड़े पैमाने पर चर्चा की गई है)। कई हालिया माइक्रोप्रोटीन पहचान अध्ययन 38,39,40,41,42,43,44,45,46,47 ने फाइलोस्सफ 48,49 नामक एक ऐसे एल्गोरिदम के उपयोग पर बहुत अधिक भरोसा किया है , एक शक्तिशाली तुलनात्मक जीनोमिक्स दृष्टिकोण जिसका लाभ जीनोम के संरक्षित प्रोटीन-कोडिंग क्षेत्रों को उन लोगों से अलग करने के लिए किया जा सकता है जो नॉनकोडिंग हैं।
फिलोस्सफ प्रोटीन-कोडिंग जीन के विकासवादी हस्ताक्षरों का पता लगाने के लिए बहु-प्रजाति न्यूक्लियोटाइड संरेखण और फाइटोलैनेटिक मॉडल का उपयोग करके कोडन प्रतिस्थापन आवृत्तियों (सीएसएफ) की तुलना करता है। यह अनुभवजन्य मॉडल-आधारित दृष्टिकोण इस आधार पर निर्भर करता है कि प्रोटीन मुख्य रूप से न्यूक्लियोटाइड अनुक्रम के बजाय अमीनो एसिड स्तर पर संरक्षित होते हैं। इसलिए, पर्यायवाची कोडन प्रतिस्थापन, जो संरक्षित गुणों (यानी, चार्ज, हाइड्रोफोबिसिटी, ध्रुवीयता) के साथ अमीनो एसिड के लिए एक ही अमीनो एसिड, या कोडन प्रतिस्थापन को एन्कोड करते हैं, सकारात्मक रूप से स्कोर किए जाते हैं, जबकि मिसेंस और बकवास प्रतिस्थापन सहित गैर-पर्यायवाची प्रतिस्थापन नकारात्मक स्कोर करते हैं। फिलोसीएसएफ को पूरे जीनोम डेटा पर प्रशिक्षित किया जाता है और पूर्ण अनुक्रम से अलगाव में कोडिंग अनुक्रम (सीडीएस) के छोटे हिस्सों को स्कोर करने में प्रभावी साबित हुआ है, जो मानक प्रोटीन-कोडिंग जीन48,49 के माइक्रोप्रोटीन या व्यक्तिगत एक्सॉन का विश्लेषण करते समय आवश्यक है।
विशेष रूप से, कैलिफोर्निया सांताक्रूज विश्वविद्यालय (यूसीएससी) जीनोम ब्राउज़र 49,50,51 में फिलोस्सफ ट्रैक हबका हालिया एकीकरण सभी पृष्ठभूमि के जांचकर्ताओं को प्रोटीन-कोडिंग क्षमता के लिए ब्याज के जीनोमिक क्षेत्रों को क्वेरी करने के लिए उपयोगकर्ता के अनुकूल इंटरफ़ेस तक आसानी से पहुंचने में सक्षम बनाता है। नीचे उल्लिखित प्रोटोकॉल यूसीएससी जीनोम ब्राउज़र पर फिलोसीएसएफ ट्रैक हब लोड करने के तरीके पर विस्तृत निर्देश प्रदान करता है और बाद में उच्च आत्मविश्वास वाले प्रोटीन-कोडिंग क्षेत्रों (या इसकी कमी) की जांच करने के लिए ब्याज के जीनोमिक क्षेत्रों से पूछताछ करता है। इसके अतिरिक्त, उस मामले में जहां एक सकारात्मक फिलोस्सफ स्कोर मनाया जाता है, माइक्रोप्रोटीन-कोडिंग क्षमता का विश्लेषण करने और क्रॉस-प्रजाति अनुक्रम संरक्षण को चित्रित करने के लिए पहचाने गए अमीनो एसिड अनुक्रमों के कई प्रजातियों के संरेखण को कुशलतापूर्वक उत्पन्न करने के लिए चरणों को चित्रित किया जाता है। अंत में, कई अतिरिक्त सार्वजनिक रूप से उपलब्ध संसाधनों और उपकरणों को पहचाने गए माइक्रोप्रोटीन विशेषताओं का सर्वेक्षण करने के लिए चर्चा में पेश किया जाता है, जिसमें भविष्यवाणी की गई डोमेन संरचनाएं और पुटेटिव माइक्रोप्रोटीन फ़ंक्शन में अंतर्दृष्टि शामिल है।
Subscription Required. Please recommend JoVE to your librarian.
Protocol
प्रोटोकॉल यूसीएससी जीनोम ब्राउज़र (मुज एट अल द्वारा उत्पन्न) पर फिलोस्सएफ ब्राउज़र ट्रैक को लोड करने और नेविगेट करने के लिए विवरण चरणों के नीचे उल्लिखित है। यूसीएससी जीनोम ब्राउज़र के बारे में सामान्य प्रश्नों के लिए, एक व्यापक जीनोम ब्राउज़र उपयोगकर्ता गाइड यहां पाया जा सकता है: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html।
1. यूसीएससी जीनोम ब्राउज़र में फिलोस्सफ ट्रैक हब लोड हो रहा है
- एक इंटरनेट ब्राउज़र विंडो खोलें और यूसीएससी जीनोम ब्राउज़र (https://genome.ucsc.edu/) पर नेविगेट करें।
- हमारे उपकरण शीर्षक के तहत, हब ट्रैक करें विकल्प का चयन करें।
नोट:: ट्रैक हब विकल्प भी मेरा डेटा टैब के अंतर्गत पाया जा सकता है। - सार्वजनिक हब टैब में, खोज शब्द बॉक्स में फ़ाइलोस्सफ़ टाइप करें। सर्च पब्लिक हब बटन पर क्लिक करें।
- हब नाम फिलोस्सएफ के लिए कनेक्ट बटन पर क्लिक करके फिलोस्सफ से कनेक्ट करें (विवरण: विकासवादी प्रोटीन-कोडिंग क्षमता जैसा कि फिलोस्सफ द्वारा मापा गया है)।
नोट: यह ट्रैक हब मानव (एचजी 1 9 और एचजी 38) और माउस (मिमी 10 और मिमी 3 9) सहित कई विधानसभाओं में लोड होगा। - कनेक्ट पर क्लिक करने के बाद, यूसीएससी जीनोम ब्राउज़र गेटवे पेज (https://genome.ucsc.edu/cgi-bin/hgGateway) पर रीडायरेक्ट होने की प्रतीक्षा करें।
2. जीन पहचानकर्ताओं का उपयोग करके ब्याज के जीन पर नेविगेट करना
- क्वेरी करने के लिए प्रजातियों और जीनोम असेंबली का चयन करें। किसी भिन्न प्रजाति (जैसे, माउस) से पूछताछ करने के लिए, उपयुक्त आइकन पर क्लिक करके ब्राउज़ /प्रजातियों का चयन करें शीर्षक के तहत रुचि की प्रजातियों का चयन करें , या प्रजातियों को टेक्स्ट बॉक्स में टाइप करें जो कहता है, प्रजातियां, सामान्य नाम या असेंबली आईडी दर्ज करें।
नोट:: असेंबली सीधे स्थिति ढूँढें शीर्षक के अंतर्गत सूचीबद्ध है। आमतौर पर, डिफ़ॉल्ट मानव असेंबली है (उदाहरण के लिए, दिसंबर 2009 [GRCh37 / - ड्रॉपडाउन मेनू का उपयोग करके स्थिति ढूँढें शीर्षक के तहत खोज करने के लिए असेंबली चुनें।
- खोज शब्द बॉक्स में स्थिति, जीन प्रतीक, या खोज शब्द दर्ज करें और जीनोम ब्राउज़र पर रुचि के जीन पर नेविगेट करने के लिए जाओ पर क्लिक करें।
- यदि खोज के परिणामस्वरूप कई मिलान हुए हैं, तो उस पृष्ठ पर रीडायरेक्ट होने की प्रतीक्षा करें जिसमें रुचि की स्थिति के चयन की आवश्यकता होती है। ब्याज के उपयुक्त जीन पर क्लिक करें।
3. अनुक्रम जानकारी का उपयोग करके ब्याज के जीनोमिक क्षेत्रों में नेविगेट करना
- यूसीएससी जीनोम ब्राउज़र (https://genome.ucsc.edu/) पर नेविगेट करें और एक विशिष्ट डीएनए या प्रोटीन अनुक्रम से पूछताछ करने के लिए हमारे टूल शीर्षक के तहत ब्लास्ट-लाइक संरेखण टूल (बीएलएटी) का चयन करें। वैकल्पिक रूप से, कर्सर को टूल टैब पर होवर करें और ब्लैट विकल्प का चयन करें या इस लिंक का पालन करें: https://genome.ucsc.edu/cgi-bin/hgBlat।
- ड्रॉपडाउन मेनू का उपयोग करके प्रजातियों (जीनोम) और ब्याज की विधानसभा का चयन करें।
- ड्रॉपडाउन मेनू का उपयोग करके क्वेरी प्रकार परिभाषित करें।
- रुचि का अनुक्रम ब्लैट खोज जीनोम पाठ बॉक्स में चिपकाएँ और सबमिट करें क्लिक करें.
- ब्याज के जीनोमिक क्षेत्र में नेविगेट करने के लिए एक्शन हेडिंग के तहत ब्राउज़र लिंक पर क्लिक करें।
4. फिलोस्सफ ट्रैक डेटा का उपयोग करके संरक्षित एसओआरएफ की पहचान करना
- नेत्रहीन सकारात्मक रूप से स्कोरिंग फिलोस्सफ क्षेत्रों (चित्रा 1) के लिए ब्याज के जीनोमिक क्षेत्र को स्कैन करें।
नोट: यूसीएससी जीनोम ब्राउज़र पर फिलोस्सफ स्कोर की नेत्रहीन व्याख्या करने के तरीके के विस्तृत विवरण के लिए, नीचे दिए गए प्रतिनिधि परिणाम अनुभाग देखें। - अनुक्रम विशेषताओं की जांच करने और स्टार्ट / स्टॉप कोडन की खोज करने के लिए ब्याज के क्षेत्रों को बढ़ाने के लिए ज़ूम सुविधा का उपयोग करें। मैन्युअल रूप से ज़ूम इन करने के लिए, शिफ्ट कुंजी दबाए रखें और ब्याज के क्षेत्र के साथ खींचते समय माउस बटन पर क्लिक करें और दबाए रखें। वैकल्पिक रूप से, नेविगेट करने के लिए पृष्ठ के शीर्ष पर ज़ूम इन और ज़ूम आउट बटन का उपयोग करें (1.5x, 3x, 10x, या बेस ज़ूम विकल्प उपलब्ध हैं)।
नोट: ज़ूम इन/ज़ूम आउट बटन का उपयोग करने से पहले, जीन को पुनर्स्थापित करना आवश्यक है ताकि ब्याज का क्षेत्र स्क्रीन के बीच में हो। इस क्रिया को करने के लिए, छवि पर क्लिक करें और जीनोमिक क्षेत्र को क्षैतिज रूप से वांछित रूप से स्थानांतरित करने के लिए इसे बाएं या दाएं खींचें या पृष्ठ के शीर्ष पर चाल तीर का उपयोग करें। - न्यूक्लियोटाइड (बेस) अनुक्रम दिखाई देने तक ज़ूम इन करें।
नोट: न्यूक्लियोटाइड अनुक्रम सीधे +1 चिकना फिलोस्सएफ स्कोर के ऊपर दिखाई देगा। - नेत्रहीन सकारात्मक स्कोरिंग फिलोस्सएफ क्षेत्रों की शुरुआत और अंत के पास न्यूक्लियोटाइड अनुक्रम को स्कैन करें ताकि पुटेटिव स्टार्ट (एटीजी) और स्टॉप (टीजीए / टीएए / टीएजी) कोडन की पहचान की जा सके।
नोट: यदि ब्याज का जीन डीएनए के माइनस स्ट्रैंड पर है, तो स्टार्ट और स्टॉप कोडन रिवर्स पूरक होगा (यानी, स्टार्ट कोडन के लिए कैट और स्टॉप कोडन के लिए टीसीए /
5. अन्य जीनोम में समरूप क्षेत्रों को देखना
- पृष्ठ के शीर्ष पर दृश्य शीर्ष पर माउस को घुमाएं और अन्य जीनोम (कन्वर्ट) विकल्प पर क्लिक करें।
- न्यू जीनोम शीर्षक के नीचे ड्रॉपडाउन मेनू का उपयोग करके ब्याज के जीनोम को परिभाषित करें।
- नई असेंबली शीर्षक के तहत ड्रॉपडाउन मेनू का उपयोग करके ब्याज की जीनोमिक असेंबली का चयन करें, फिर सबमिट बटन पर क्लिक करें।
- एक बार जब ब्राउज़र समानता के साथ नई असेंबली में क्षेत्रों की एक सूची लौटाता है, तो ब्याज के समरूप क्षेत्र में नेविगेट करने के लिए गुणसूत्र स्थिति लिंक पर क्लिक करें।
नोट: कुल ठिकानों (न्यूक्लियोटाइड) का प्रतिशत और क्षेत्र द्वारा कवर किए गए स्पैन को सूचीबद्ध प्रत्येक क्षेत्र के लिए परिभाषित किया जाएगा। मिलान करने वाले ठिकानों का प्रतिशत जितना अधिक होगा, ब्याज के क्षेत्र के लिए संरक्षण उतना ही अधिक होगा। - अनुक्रम का विश्लेषण करने के लिए धारा 4 में विस्तृत एक ही नौवहन रणनीतियों का पालन करें।
6. ब्याज के माइक्रोप्रोटीन के लिए बहु-प्रजाति अनुक्रम संरेखण उत्पन्न करना
- जीन विवरण पृष्ठ पर नेविगेट करने के लिए यूसीएससी जीनोम ब्राउज़र (नीले बॉक्स के साथ चित्रा 1 ए में इंगित) पर जेनकोड ट्रैक में रुचि के जीन पर क्लिक करें।
- उपकरण और डेटाबेस के अनुक्रम और लिंक शीर्षक के तहत, तालिका में लिंक पर क्लिक करें जो अन्य प्रजाति फास्टा पढ़ता है।
- उन्हें चुनने के लिए ब्याज की प्रजातियों से जुड़े बक्से पर क्लिक करें। सबमिट बटन पर क्लिक करें। फास्टा प्रारूप में पृष्ठ के निचले भाग में दिखाई देने वाले अनुक्रमों को वर्ड प्रोसेसिंग दस्तावेज़ में कॉपी और पेस्ट करें।
- एक दूसरी ब्राउज़र विंडो खोलें और यूरोपीय जैव सूचना विज्ञान संस्थान (ईएमबीएल-ईबीआई) वेबसाइट53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/ पर क्लस्टल ओमेगा मल्टीपल सीक्वेंस संरेखण टूल 52 पर नेविगेट करें।
- अनुक्रम फ़ाइलें जो अभी भी क्लिपबोर्ड पर हैं चरण 1 में बॉक्स में चिपकाएँ जो किसी भी समर्थित स्वरूप में अनुक्रम पढ़ता है। पृष्ठ के निचले भाग में स्क्रॉल करें और सबमिट पर क्लिक करें। प्रतीकों के लिए संरेखित परिणाम (काले फ़ॉन्ट में) के नीचे देखें जो प्रत्येक अमीनो एसिड के संरक्षण की डिग्री को इंगित करते हैं (प्रतीकों को तालिका 1 में परिभाषित किया गया है)।
नोट: संरेखण उत्पन्न करने के लिए कई मिनट लग सकते हैं। - अमीनो एसिड गुणों को रंग में देखने के लिए, अमीनो एसिड को उनके गुणों के अनुसार रंगने के लिए अनुक्रमों के ऊपर सीधे रंग दिखाएं लिंक पर क्लिक करें ( तालिका 2 में परिभाषित)।
- एक आंकड़ा या चित्रण फ़ाइल उत्पन्न करने के लिए एक शब्द प्रसंस्करण या स्लाइड शो प्रोग्राम में अनुक्रम संरेखण को कॉपी और पेस्ट करें (उदाहरण के लिए, चित्रा 2)।
नोट: कूरियर जैसे संरेखण के लिए एक मोनोस्पेस फ़ॉन्ट का उपयोग करें। - क्लूस्टल ओमेगा परिणाम पृष्ठ से अन्य आउटपुट देखने के लिए, उपयुक्त टैब (यानी, गाइड ट्री या फाइलोजेनेटिक ट्री) पर क्लिक करें।
- जलव्यू का उपयोग करके अनुक्रम जानकारी देखने के विकल्पों के लिए परिणाम दर्शक टैब पर क्लिक करें, एक मुफ्त प्रोग्राम जो कई अनुक्रम संरेखण संपादन, विज़ुअलाइज़ेशन और विश्लेषण55 में माहिर है, या एमव्यू और सिंपल फिलोजेनी56 के सीधे लिंक तक पहुंचने के लिए।
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
यहां हम मान्य माइक्रोप्रोटीन माइटोरेगुलिन (एमटीएलएन) का उपयोग एक उदाहरण के रूप में करेंगे ताकि यह प्रदर्शित किया जा सके कि कैसे एक संरक्षित एसओआरएफ एक सकारात्मक फिलोसीएसएफ स्कोर उत्पन्न करेगा जिसे यूसीएससी जीनोम ब्राउज़र पर आसानी से कल्पना और विश्लेषण किया जा सकता है। मिटोरेगुलिन को पहले एक नॉनकोडिंग आरएनए (पूर्व में मानव जीन आईडी एलआईएनसी00116 और माउस जीन आईडी 1500011के16आरआईके) के रूप में एनोटेट किया गया था। तुलनात्मक जीनोमिक्स और अनुक्रम संरक्षण विश्लेषण विधियों ने इन विधियों की ताकत को उजागर करते हुए अपनी प्रारंभिक खोज 40,57,58,59,60,61 में महत्वपूर्ण भूमिका निभाई। इस उदाहरण के लिए, माउस GRCm38/mm10 (दिसम्बर 2011) असेंबली का उपयोग किया जाएगा। खोज प्रोटोकॉल अनुभाग 2 में वर्णित जीन पहचानकर्ताओं (माइटोरेगुलिन, एमटीएलएन) या जीन स्थिति (सीएचआर 2: 127,791,364-127,792,496) का उपयोग करके की जा सकती है। वैकल्पिक रूप से, माइटोरेगुलिन के लिए अमीनो एसिड अनुक्रम (चित्रा 2 में दिखाया गया है) को बीएलएटी टूल (प्रोटोकॉल अनुभाग 3 में वर्णित) का उपयोग करके खोजा जा सकता है।
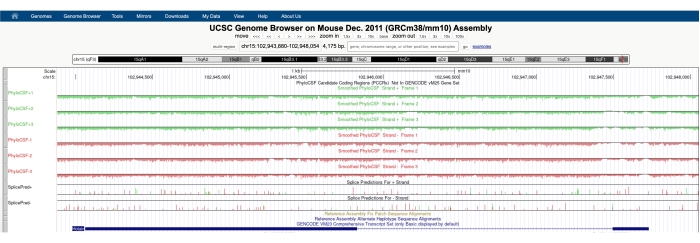
चित्रा 1 ए में दर्शाए गए एक के समान एक स्क्रीन स्क्रीन के शीर्ष पर दिखाई देने वाले फिलोस्सफ ट्रैक हब के साथ दिखाई देगी। चिकना फिलोस्सएफ पटरियों (एक छिपे हुए मार्कोव मॉडल के साथ चिकना किया गया है जो एक संभावना को परिभाषित करता है कि प्रत्येक कोडन कोडिंग है) को छह कुल पटरियों के रूप में चित्रित किया गया है, डीएनए के प्लस स्ट्रैंड के अनुरूप तीन ट्रैक (हरे रंग में फिलोस्सफ +1, +2 और +3 के रूप में चित्रित) और डीएनए के माइनस स्ट्रैंड के अनुरूप तीन ट्रैक (लाल रंग में फिलोस्सफ -1 के रूप में चित्रित, -2 और -3)। ये ट्रैक प्रत्येक दिशा में ब्याज के जीन के लिए तीन संभावित पढ़ने के फ्रेम का प्रतिनिधित्व करते हैं। ब्राउज़र विंडो पर, एक्सॉन को पतली नीली क्षैतिज रेखाओं से जुड़े नीले आयतों के रूप में दर्शाया गया है, जो इंट्रॉन का प्रतिनिधित्व करते हैं। इंट्रोनिक क्षेत्रों पर तीरहेड्स इंगित करते हैं कि जीन को किस दिशा में स्थानांतरित किया गया है (और इस प्रकार, फिलोस्सफ स्कोर के लिए किस स्ट्रैंड पर ध्यान केंद्रित करना है)। चित्रा 1 में एमटीएलएन के उदाहरण के लिए, इंट्रोनिक एरोहेड बाईं ओर इशारा कर रहे हैं। इसलिए, एमटीएलएन जीन को डीएनए के माइनस स्ट्रैंड से ट्रांसक्रिप्ट किया गया है, और प्रासंगिक फिलोस्सएफ स्कोर को -1, -2 और -3 ट्रैक (लाल रंग में) में दर्शाया गया है।
प्रत्येक फिलोसीएसएफ ट्रैक को एक पतली काली रेखा के रूप में दर्शाया गया है जिसमें नकारात्मक स्कोरिंग क्षेत्रों को रेखा के नीचे हल्के हरे / लाल रंग में दर्शाया गया है और सकारात्मक स्कोरिंग क्षेत्रों को रेखा के ऊपर गहरे हरे / लाल रंग में दर्शाया गया है। जैसा कि परिचय में वर्णित है, एक सकारात्मक फिलोस्सफ स्कोर एक संरक्षित क्षेत्र को इंगित करता है जो संभवतः कोडिंग है। ध्यान दें कि विशेष रूप से उच्च अनुक्रम संरक्षण वाले प्रोटीन-कोडिंग क्षेत्रों के लिए, वे अक्सर एंटीसेंस स्ट्रैंड पर सकारात्मक स्कोर करते हैं; हालाँकि, फिलोसीएसएफ स्कोर आमतौर पर सही स्ट्रैंड पर अधिक होता है। उदाहरण के लिए, यह एमटीएलएन के लिए चित्रा 1 में देखा जा सकता है जहां सही कोडिंग अनुक्रम फाइलोस्स्फ -1 ट्रैक में बहुत अधिक स्कोर करता है, और एंटीसेंस स्ट्रैंड (फिलोस्सफ +2 ट्रैक) भी एक सकारात्मक स्कोर उत्पन्न करता है। जैसा कि चित्रा 1 ए (ब्लैक बॉक्स के साथ इंगित) में देखा गया है, एमटीएलएन के पहले एक्सॉन में एक क्षेत्र है जो फिलोस्सफ -1 ट्रैक पर बहुत अधिक स्कोर करता है, यह सुझाव देता है कि यह एक कोडिंग क्षेत्र के अनुरूप हो सकता है। इस क्षेत्र को और विस्तार से जांचने के लिए, क्षेत्र (चित्रा 1 बी) को ज़ूम इन और आवर्धित करना सहायक है। जैसा कि चित्रा 1 सी, डी में दिखाया गया है, एमटीएलएन के पहले एक्सॉन में सकारात्मक स्कोरिंग क्षेत्र सीधे स्टार्ट कोडन (चित्रा 1 सी) पर शुरू होता है और स्टॉप कोडन (चित्रा 1 डी) पर समाप्त होता है, जो इंगित करता है कि यह ओआरएफ अत्यधिक संरक्षित है और दृढ़ता से सुझाव देता है कि यह एक कोडिंग ओआरएफ है। चूंकि एमटीएलएन डीएनए के माइनस स्ट्रैंड पर है, स्टार्ट और स्टॉप कोडन को कोडन के रिवर्स पूरक के रूप में दिखाया गया है (यानी, एटीजी स्टार्ट कोडन को कैट [चित्रा 1 सी] के रूप में दिखाया गया है और टीजीए स्टॉप कोडन को टीसीए [चित्रा 1 डी]) के रूप में दिखाया गया है।
माइक्रोप्रोटीन-कोडिंग क्षमता वाले संरक्षित क्षेत्रों की खोज के लिए फिलोस्सफ का उपयोग करने के अलावा, इस तकनीक को संरक्षित ओआरएफ की उपस्थिति को खारिज करने के लिए नॉनकोडिंग आरएनए के पहले-पास विश्लेषण के रूप में भी लागू किया जा सकता है, इस प्रकार एक नॉनकोडिंग एनोटेशन के लिए समर्थन प्रदान करता है। उदाहरण के लिए, फिलोस्सफ का उपयोग करके अच्छी तरह से विशेषता वाले एलएनसीआरएनए हॉटएयर 62,63 का विश्लेषण सभी छह पटरियों (चित्रा 3) में पूरे जीन में एक नकारात्मक स्कोर दिखाता है, जो अनुक्रम संरक्षण की कमी का दृढ़ता से संकेत देता है और समर्थन प्रदान करता है कि हॉटएयर को सही ढंग से एक नॉनकोडिंग आरएनए के रूप में एनोटेट किया गया है।
जैसा कि चित्रा 1 में स्पष्ट रूप से देखा गया है, माइटोरेगुलिन के लिए संपूर्ण कोडिंग ओआरएफ एक एकल एक्सॉन के भीतर स्थित है, जिससे एक एकल, निर्बाध, सकारात्मक स्कोरिंग क्षेत्र के साथ फिलोस्सफ द्वारा एक सरल और सीधा रीडआउट का उत्पादन होता है। हालांकि, फिलोस्सफ ट्रैक हब डेटा हमेशा स्पष्ट और व्याख्या करने में आसान नहीं होता है। उदाहरण के लिए, माउस 1810058I24Rik जीन 47,64,65 द्वारा एन्कोडेड माइटोलम्बन / Stmp1/MM47 माइक्रोप्रोटीन एक संरक्षित ओआरएफ को दर्शाता है जो तीन एक्सॉन (चित्रा 4A) तक फैला हुआ है, और सकारात्मक फिलोस्सफ़ स्कोर एक्सॉन 1 (चित्रा 4B) में +2 ट्रैक से एक्सॉन 2 (चित्रा 4C) में +3 ट्रैक तक कूदता है, और फिर वापस + 2 ट्रैक पर वापस आता है ). जबकि पहली नज़र में यह भ्रामक दिखता है, स्पष्टीकरण काफी सीधा है। इंट्रॉन आर्किटेक्चर पर विचार किए बिना जीनोमिक क्षेत्रों के छह संभावित रीडिंग फ्रेम (डीएनए के प्लस स्ट्रैंड पर तीन और माइनस स्ट्रैंड पर तीन) स्कोर करता है। इसलिए, यह पढ़ने के फ्रेम की 3-न्यूक्लियोटाइड आवधिकता में इंट्रोनिक अनुक्रम जानकारी को बरकरार रखता है। इस प्रकार, यदि एक इंट्रॉन में कई न्यूक्लियोटाइड होते हैं जो तीन (यानी, तीन न्यूक्लियोटाइड / कोडन) से विभाज्य नहीं होते हैं, तो फिलोस्सफ रीडिंग फ्रेम एक ट्रैक से दूसरे ट्रैक पर कूद जाएगा।
अंत में, फिलोसीएसएफ का उपयोग एक आरएनए अणु के भीतर कई अलग-अलग कोडिंग ओआरएफ की पहचान करने के लिए प्रभावी ढंग से किया जा सकता है। उदाहरण के लिए, एमआईईएफ 1 माइक्रोप्रोटीन (एमआईईएफ 1-एमपी) माइटोकॉन्ड्रियल बढ़ाव कारक 1 (एमआईईएफ 1) 66 (चित्रा 5) के 5 'यूटीआर के भीतर एन्कोडेड है। जब एमआईईएफ 1 जीनोमिक क्षेत्र का विश्लेषण फिलोस्सफ द्वारा किया जाता है, तो एमआईईएफ 1-एमपी (चित्रा 5 सी) के अनुरूप एक असतत सकारात्मक फिलोस्सएफ स्कोर को एमआईईएफ 1 (चित्रा 5 बी) के लिए मुख्य सीडीएस के ऊपर आसानी से देखा जा सकता है। एमआईईएफ 1 और उससे जुड़े माइक्रोप्रोटीन (एमआईईएफ 1-एमपी) पर आगे की चर्चा इस लेख में उल्लिखित विधियों और प्रोटोकॉल की ताकत और कमजोरियों के सारांश के साथ चर्चा में नीचे दी गई है।

चित्रा 1: माइटोरेगुलिन (एमटीएलएन) जीन का फिलोसीएसएफ विश्लेषण एक मान्य माइक्रोप्रोटीन के अनुरूप उच्च अनुक्रम संरक्षण के एक क्षेत्र को इंगित करता है। (ए) यूसीएससी जीनोम ब्राउज़र और फिलोस्सफ ट्रैक्स के स्क्रीनशॉट से पता चलता है कि एमटीएलएन में दो एक्सॉन और एक एकल इंट्रॉन होता है। बाईं ओर इंट्रॉन बिंदु के भीतर तीरहेड्स, यह दर्शाता है कि एमटीएलएन जीन डीएनए के माइनस स्ट्रैंड से स्थानांतरित किया गया है, और प्रासंगिक फिलोसीएसएफ स्कोर इसलिए -1, -2 और -3 ट्रैक (लाल रंग में) में दिखाए गए हैं। पूर्ण माइटोरेगुलिन कोडिंग अनुक्रम एक्सॉन 1 के भीतर निहित है और फिलोस्सफ -1 ट्रैक (बी) पर अत्यधिक स्कोर करता है। फिलोस्सफ -1 ट्रैक (सी) में सकारात्मक स्कोरिंग क्षेत्र की शुरुआत में एक संरक्षित स्टार्ट कोडन स्पष्ट रूप से देखा जा सकता है, जिसे ग्रीन बॉक्स (कैट, रिवर्स पूरक एटीजी) के साथ हाइलाइट किया गया है। इसके अतिरिक्त, एक संरक्षित स्टॉप कोडन (टीसीए, रिवर्स पूरक टीजीए) को पैनल (डी) में एक लाल बॉक्स के साथ इंगित किया जाता है, जो सकारात्मक स्कोरिंग फिलोस्सएफ क्षेत्र के अंत के साथ संरेखित होता है। एमटीएलएन जीन के बारे में विस्तृत जानकारी नीले बॉक्स (पैनल ए में दिखाया गया है) के भीतर एमटीएलएन जीन पहचानकर्ता पर क्लिक करके पाई जा सकती है। ध्यान दें, अत्यधिक संरक्षित प्रोटीन-कोडिंग क्षेत्र अक्सर एंटीसेंस स्ट्रैंड पर सकारात्मक रूप से स्कोर करते हैं (यहां एमएलएन के लिए फिलोस्सफ +2 ट्रैक में देखा गया है)। हालांकि, फिलोस्सफ स्कोर आमतौर पर सही स्ट्रैंड (इस उदाहरण में फिलोस्सफ -1 ट्रैक) पर अधिक होता है। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहां क्लिक करें।

चित्रा 2: क्लस्टल ओमेगा कार्यक्रम का उपयोग करके उत्पन्न माइक्रोप्रोटीन माइटोरेगुलिन की कई प्रजातियां अनुक्रम संरेखण। संकेतित आठ प्रजातियों के लिए माइटोरेगुलिन एमिनो एसिड अनुक्रम प्रोटोकॉल अनुभाग 6 में विस्तृत के रूप में निकाले गए थे और क्लस्टल ओमेगा एकाधिक अनुक्रम संरेखण उपकरण के साथ गठबंधन किया गया था। अमीनो एसिड के गुणों को रंग (लाल, छोटे / हाइड्रोफोबिक; नीला, अम्लीय; मैजेंटा, बुनियादी; हरा, हाइड्रॉक्सल / सल्फहाइड्रिल / अमाइन) द्वारा इंगित किया जाता है (आगे तालिका 2 में परिभाषित)। अमीनो एसिड के नीचे के प्रतीक संरक्षण की डिग्री (तारांकन, पूरी तरह से संरक्षित अवशेष; बृहदान्त्र, दृढ़ता से समान गुणों वाले अमीनो एसिड; अवधि, कमजोर समान गुणों के समूहों के बीच संरक्षण) ( तालिका 1 में बड़े पैमाने पर विस्तृत) को इंगित करते हैं। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहां क्लिक करें।
चित्रा 3: मान्य लंबे नॉनकोडिंग आरएनए हॉटेयर के लिए फिलोस्सफ पटरियों का एक स्क्रीनशॉट अपने जीनोमिक लोकस में अनुक्रम संरक्षण की कमी दिखाता है। हॉटेयर के इंट्रोनिक क्षेत्र में एरोहेड्स बाईं ओर इशारा कर रहे हैं, यह दर्शाता है कि एलएनसीआरएनए डीएनए के नकारात्मक स्ट्रैंड से स्थानांतरित किया गया है, और इसलिए फिलोसीएसएफ -1, -2 और -3 ट्रैक विश्लेषण का फोकस होना चाहिए। ध्यान दें कि फिलोसीएसएफ स्कोर पूरे जीन (सभी छह पटरियों के लिए) में नकारात्मक है, जो अनुक्रम संरक्षण की कमी का संकेत देता है, जो नॉनकोडिंग आरएनए के रूप में इसके उचित एनोटेशन का समर्थन करता है। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहां क्लिक करें।

चित्रा 4: माउस 1810058I24Rik जीन का फिलोसीएसएफ विश्लेषण, जो माइक्रोप्रोटीन माइटोलम्बन / Stmp1/ MM47 को एन्कोड करता है। (A) माउस 1810058I24Rik जीन में तीन एक्सॉन शामिल हैं, और इंट्रोनिक क्षेत्रों में एरोहेड्स दाईं ओर इंगित करते हैं, यह दर्शाता है कि यह डीएनए के प्लस स्ट्रैंड पर ट्रांसक्रिप्ट किया गया है और इसलिए फिलोसीएसएफ + 1, +2, और +3 ट्रैक संरक्षित माइक्रोप्रोटीन कोडिंग अनुक्रम सभी तीन एक्सॉन को फैलाता है, एक्सॉन 1 (बी) में शुरू होता है, एक्सॉन 2 (सी) के माध्यम से पढ़ता है, और एक्सॉन 3 (डी) में समाप्त होता है। ध्यान दें कि सकारात्मक फिलोसीएसएफ स्कोर एक्सॉन 1 में +2 ट्रैक, एक्सॉन 2 में +3 ट्रैक और एक्सॉन 1 में +2 ट्रैक पर पाया जाता है। एक ट्रैक से दूसरे ट्रैक तक सकारात्मक स्कोर के आंदोलन का कारण यह है कि फिलोसीएसएफ जीन के एक्सॉन / इंट्रॉन संरचना से स्वतंत्र डीएनए अनुक्रम के छह संभावित रीडिंग फ्रेम का विश्लेषण करता है। इसलिए, कई न्यूक्लियोटाइड युक्त एक इंट्रॉन जो तीन (तीन न्यूक्लियोटाइड / कोडन) से विभाज्य नहीं है, रीडिंग फ्रेम में एक अलग ट्रैक पर बदलाव का कारण होगा। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहां क्लिक करें।

चित्रा 5: फिलोस्सएफ के साथ एमआईईएफ 1 जीनोमिक लोकस का विश्लेषण 5 'यूटीआर में प्रोटीन-कोडिंग क्षमता वाले क्षेत्र की पहचान करता है जो साझा आरएनए पर मुख्य एमआईईएफ 1 सीडीएस से स्वतंत्र है। इस संरक्षित अपस्ट्रीम ओआरएफ (यूओआरएफ) को एमआईईएफ 1-एमपी नामक एक माइक्रोप्रोटीन को एन्कोड करने के लिए दिखाया गया है। (ए) एमआईईएफ 1 जीनोमिक लोकस का अवलोकन। इंट्रॉन में एरोहेड दाईं ओर इंगित करते हैं, यह दर्शाता है कि एमआईईएफ 1 डीएनए के प्लस स्ट्रैंड से स्थानांतरित किया गया है (कोडिंग क्षमता निर्धारित करने के लिए फिलोस्सफ +1, +2 और +3 ट्रैक पर ध्यान केंद्रित करें)। मुख्य Mief1 सीडीएस एक 463 एमिनो एसिड प्रोटीन एन्कोड करता है और पैनल (बी) में दिखाया गया है। हालांकि, एमआईईएफ 1 के 5 'यूटीआर के भीतर एक अलग संरक्षित अपस्ट्रीम ओआरएफ भी है जो एक अद्वितीय 70 एमिनो एसिड माइक्रोप्रोटीन को एन्कोड करता है जिसे एमआईईएफ 1-एमपी (सी) कहा जाता है। जैसा कि पैनल सी में देखा गया है, एमआईईएफ 1-एमपी की एमआईईएफ 1 5 'यूटीआर के भीतर अपनी संरक्षित शुरुआत और स्टॉप कोडन है, और ओआरएफ फाइलोस्सफ +1 ट्रैक पर बहुत अधिक स्कोर करता है, मजबूत सबूत प्रदान करता है कि यह एक कार्यात्मक माइक्रोप्रोटीन को एन्कोड करता है। संक्षिप्त नाम: ओआरएफ = ओपन रीडिंग फ्रेम; यूओआरएफ = अपस्ट्रीम ओआरएफ; यूटीआर = अनुवादित क्षेत्र; सीडीएस = कोडिंग अनुक्रम। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहां क्लिक करें।
| चिह्न | अमीनो एसिड संरक्षण का स्तर | समूहीकृत अमीनो एसिड |
| तारांकन (*) | पूरी तरह से संरक्षित अवशेष | लागू नहीं (एकल, पूरी तरह से संरक्षित अवशेष) |
| बृहदान्त्र (:) | दृढ़ता से समान गुणों वाले समूह | एसटीए; एनईक्यूके; एनएचक्यूके; एनडीईक्यू; क्यूएचआरके; एमआईएलवी; एमआईएलए; एचवाई; एफवाईडब्ल्यू |
| अवधि (.) | कमजोर समान गुणों वाले समूह | सीएसए; एटीवी; एसएजी; एसटीएनके; एसटीपीए; एसजीएनडी; एसएनडीईक्यूके; एनडीईक्यूएचके; एनईक्यूएचआरके; एफवीएलआईएम; एचएफवाई |
| अंतरिक्ष (कोई प्रतीक नहीं) | कोई समानता नहीं | लागू नहीं है (कोई समानता नहीं) |
तालिका 1: क्लूस्टल ओमेगा द्वारा उत्पन्न एकाधिक अनुक्रम संरेखण के लिए सर्वसम्मति प्रतीकों की परिभाषाएं। चित्रा 2 में दिखाए गए कई प्रजातियों के अनुक्रम संरेखण को क्लस्टल ओमेगा52 का उपयोग करके उत्पन्न किया गया था। संक्षिप्त नाम: सेरीन (एस), थ्रेओनिन (टी), एलानिन (ए), शतावरी (एन), ग्लूटामिक एसिड (ई), ग्लूटामाइन (क्यू), लाइसिन (के), एस्पार्टिक एसिड (डी), आर्जिनिन (आर), मेथियोनीन (एम), आइसोल्यूसीन (आई), ल्यूसीन (एल), फेनिलएलनिन (एफ), हिस्टिडीन (एच), टायरोसिन (वाई), ट्रिप्टोफैन (डब्ल्यू),
| फ़ॉन्ट रंग | जायदाद | अमीनो एसिड अवशेष [संक्षिप्त नाम] |
| लाल | छोटे, हाइड्रोफोबिक | एलानिन [ए], वेलिन [वी], फेनिलएलनिन [एफ], प्रोलाइन [पी], मेथियोनीन [एम], आइसोल्यूसीन [आई], ल्यूसीन [एल], ट्रिप्टोफैन [डब्ल्यू] |
| नीला | अम्लीय | एसपारटिक एसिड [डी], ग्लूटामिक एसिड [ई] |
| मैजंटा | बुनियादी | आर्जिनिन [आर], लाइसिन [के] |
| हरा | हाइड्रॉक्सल, सल्फहाइड्रील, अमाइन, +जी | सेरीन [एस], थ्रेओनिन [टी], टायरोसिन [वाई], हिस्टिडीन [एच], सिस्टीन [सी], शतावरी [एन], ग्लाइसिन [जी], ग्लूटामाइन [क्यू] |
तालिका 2: चित्रा 2 में दर्शाए गए अमीनो एसिड के गुण। क्लस्टल ओमेगा52 चित्रा 2 में दिखाए गए कई अनुक्रम संरेखण उत्पन्न करने के लिए इस्तेमाल किया गया था।
Subscription Required. Please recommend JoVE to your librarian.
Discussion
यहां प्रस्तुत प्रोटोकॉल उपयोगकर्ता के अनुकूल यूसीएससी जीनोम ब्राउज़र 48,49,50,51 पर फिलोस्सफ का उपयोग करके माइक्रोप्रोटीन-कोडिंग क्षमता के लिए ब्याज के जीनोमिक क्षेत्रों से पूछताछ करने के बारेमें विस्तृत निर्देश प्रदान करता है। जैसा कि ऊपर विस्तृत है, फिलोस्सएफ एक शक्तिशाली तुलनात्मक जीनोमिक्स एल्गोरिथ्म है जो विकासवादी हस्ताक्षरों की पहचान करने के लिए फाइटोलैनेटिक मॉडल और कोडन प्रतिस्थापन आवृत्तियों को एकीकृत करता है जो प्रोटीन-कोडिंग जीन48,49 के विशिष्ट हैं। फाइलोसीएसएफ का व्यापक रूप से जीनोमिक क्षेत्रों में कार्यात्मक माइक्रोप्रोटीन की पहचान करने के लिए उपयोग किया जाता है, जिसे पहले नॉनकोडिंग 38,39,40,41,42,43,44,45,46,47 के रूप में एनोटेट किया गया था , और इस दृष्टिकोण को छोटे अनुक्रमों के लिए अन्य तुलनात्मक जीनोमिक्स विधियों को मात देने के लिए दिखाया गया है जैसे कि माइक्रोप्रोटीन 13 अमीनो एसिड के रूप में छोटे और विहित प्रोटीन के छोटे एक्सॉन के लिए 35,48,49। विशेष रूप से, विकासवादी संरक्षण के माध्यम से कार्यात्मक प्रोटीन-कोडिंग अनुक्रमों की पहचान करने के लिए एक मजबूत विधि के रूप में फिलोसीएसएफ की उपयोगिता कशेरुक और अकशेरुकी प्रजातियों से परे फैली हुई है और यहां तक कि हाल ही में सार्स-सीओवी-2 जीनोम67 की प्रोटीन-कोडिंग क्षमता से सफलतापूर्वक पूछताछ करने के लिए वायरल जीनोम पर भी लागू किया गया है।
एनोटेट नॉनकोडिंग आरएनए के भीतर पुटेटिव कोडिंग अनुक्रमों की पहचान करने के अलावा, फिलोसीएसएफ का एक फायदा यह है कि यह विहित प्रोटीन कोडिंग जीन के एनोटेट अनूदित क्षेत्रों (यूटीआर) के भीतर ओआरएफ द्वारा एन्कोडेड संरक्षित माइक्रोप्रोटीन का मज़बूती से पता लगा सकता है, जिसमें क्रमशः 5 'अपस्ट्रीम और 3' डाउनस्ट्रीम ओआरएफ (यूओआरएफ और डीओआरएफ) दोनों शामिल हैं) 8,19,66,68 . उदाहरण के लिए, एमआईईएफ 1 माइक्रोप्रोटीन (एमआईईएफ 1-एमपी) माइटोकॉन्ड्रियल बढ़ाव कारक 1 (एमआईईएफ 1) 66 के 5 'यूटीआर में एन्कोडेड है। एमआईईएफ 1-एमपी के मामले में, एमआईईएफ 1-एमपी के अनुरूप एक असतत सकारात्मक फिलोस्सएफ स्कोर ओआरएफ के ऊपर देखा जाता है जो एमआईईएफ 1 (चित्रा 5) को एन्कोड करता है। जबकि कुछ यूओआरएफ एन्कोडेड माइक्रोप्रोटीन सीधे अपने साझा एमआरएनए पर डाउनस्ट्रीम कैनोनिकल प्रोटीन के साथ बातचीत करते हैं, (उदा। इसलिए, यूओआरएफ-एन्कोडेड माइक्रोप्रोटीन की विशेषता करते समय, यह नहीं माना जाना चाहिए कि वे अपने डाउनस्ट्रीम प्रोटीन उत्पाद के प्रत्यक्ष विनियमन के माध्यम से कार्य करते हैं।
जबकि फिलोसीएसएफ में संरक्षित माइक्रोप्रोटीन-कोडिंग अनुक्रमों की पहचान के लिए एक उपकरण के रूप में कई स्पष्ट शक्तियां हैं, इस पद्धति की कई सीमाओं को पहचानना महत्वपूर्ण है। सबसे पहले, जबकि अनुक्रम संरक्षण दृढ़ता से सुझाव देता है कि एक जीनोमिक क्षेत्र कार्यात्मक चयन से गुजरा है और इस प्रकार कोडिंग है, मजबूत संरक्षण की कमी और परिणामस्वरूप नकारात्मक फिलोस्सफ स्कोर निश्चित रूप से किसी दिए गए अनुक्रम के लिए कोडिंग क्षमता से इनकार नहीं करता है। दूसरे शब्दों में, विशेष रूप से फिलोसीएसएफ पर भरोसा करने से अनुवादित ओआरएफ की निगरानी हो सकती है जो दृढ़ता से संरक्षित नहीं हैं लेकिन फिर भी कार्यात्मक माइक्रोप्रोटीन का उत्पादन करते हैं। विशेष रूप से, कम संरक्षण या नकारात्मक संरक्षण स्कोर वाले जीनोमिक क्षेत्र प्रजातियों-विशिष्ट कोडिंग क्षेत्रों या अनुक्रम विचलन या डे नोवो जीन जन्म 46,69,70,71,72,73,74 के माध्यम से विकासवादी "युवा" जीन के अनुरूप हो सकते हैं। उदाहरण के लिए, माइक्रोप्रोटीन एएसएपी, जिसे पहले मानव नॉनकोडिंग आरएनए एलआईएनसी00467 माना जाता था, द्वारा एन्कोड किया गया है, फिलोस्सफ द्वारा सकारात्मक रूप से स्कोर नहीं किया जाता है क्योंकि अमीनो एसिड अनुक्रम केवल उच्च स्तनधारियों75 में संरक्षित होता है। इसके अतिरिक्त, हाल के अध्ययनों ने कई मानव-विशिष्ट माइक्रोप्रोटीन की पहचान की, जिसमें इंटरजेनिक एलएनसीआरएनए आरपी 3-527 जी 5.1 द्वारा एन्कोडेड एक शामिल है, जो सकारात्मक फिलोस्सएफ स्कोर68,72 उत्पन्न नहीं करता है। इस संबंध में, एक सकारात्मक फिलोस्सफ स्कोर की अनुपस्थिति को एक नॉनकोडिंग क्षेत्र के प्रमाण के रूप में व्याख्या नहीं किया जा सकता है और सावधानी के साथ व्याख्या की जानी चाहिए।
फिलोस्सफ का उपयोग करते समय ध्यान में रखने के लिए एक दूसरा विचार यह है कि भले ही एक सकारात्मक स्कोर कार्यात्मक चयन और प्रोटीन-कोडिंग क्षमता का अत्यधिक विचारोत्तेजक हो, सबूत की यह पंक्ति अकेले नहीं खड़ी हो सकती है और प्रयोगात्मक रूप से मान्य होनी चाहिए। स्थिर माइक्रोप्रोटीन अभिव्यक्ति के लिए सहायक साक्ष्य उत्पन्न करने के लिए उपयोग किए जा सकने वाले तरीकों के उदाहरणों में ब्याज के माइक्रोप्रोटीन अनुक्रम के खिलाफ उठाए गए एंटीबॉडी का उपयोग करके मास स्पेक्ट्रोमेट्री या पश्चिमी सोख्ता द्वारा पुटेटिव प्रोटीन का पता लगाना शामिल है। वैकल्पिक रूप से, चूंकि इष्टतम एंटीजेनिसिटी के लिए अनुक्रम विकल्पों की कमी के कारण माइक्रोप्रोटीन के लिए विश्वसनीय एंटीबॉडी उत्पन्न करना चुनौतीपूर्ण हो सकता है, इसलिए सीआरआईएसपीआर / कैस 9 और होमोलॉजी-निर्देशित मरम्मत (एचडीआर) मार्ग का उपयोग करके पुटेटिव माइक्रोप्रोटीन अनुक्रम के साथ फ्रेम में अंतर्जात स्थान में एपिटोप टैग पेश करना भी संभव है, जिससे उच्च आत्मीयता एंटीबॉडी का उपयोग करके ब्याज के प्रोटीन का पता लगाने की सुविधा मिलती है (उदा। फ्लैग, हा, वी 5, माइक) 18. स्वीकार करने के लिए फिलोसीएसएफ की एक अंतिम सीमा यह है कि यद्यपि यह वर्तमान में आमतौर पर इस्तेमाल की जाने वाली कई जीनोमिक असेंबली में एकीकृत है, जिसमें होमो सेपियन्स (मानव एचजी 1 9, एचजी 38), मस मस्कुलस (माउस मिमी 10, मिमी 3 9), गैलस गैलस (चिकन, गैलगल 4, गैलगल 6), ड्रोसोफिला मेलानोगास्टर (फल मक्खी, डीएम 6), कैनोरहाब्डाइटिस एलिगेंस शामिल हैं (नेमाटोड, सीई11), और सार्स-सीओवी-2 (वुहकोर1), अभी भी कई प्रजातियां हैं जिन्हें वर्तमान में यूसीएससी जीनोम ब्राउज़र पर सीधे पूछताछ नहीं की जा सकती है।
पहचाने गए माइक्रोप्रोटीन के भीतर संरक्षित डोमेन या अनुक्रम विशेषताओं की पहचान उनकी कार्यात्मक प्रासंगिकता में आत्मविश्वास बढ़ाने में मदद कर सकती है और उनके पुटेटिव फ़ंक्शन में कुछ अंतर्दृष्टि प्रदान कर सकती है। यहां हम विशिष्ट उपकरणों और संसाधनों के लिए सिफारिशें प्रदान करते हैं जिनका उपयोग इस तरह की अंतर्दृष्टि प्राप्त करने के लिए आगे विस्तार से पहचाने गए माइक्रोप्रोटीन अमीनो एसिड अनुक्रमों का विश्लेषण करने के लिए किया जा सकता है। नीचे सूचीबद्ध विशिष्ट उपकरण (और सामग्री की तालिका में संक्षेप में) जनता के लिए स्वतंत्र रूप से उपलब्ध हैं, और हमने उन्हें माइक्रोप्रोटीन अध्ययन 18,38,39,40,41,47 में विशेष रूप से उपयोगकर्ता के अनुकूल और मजबूत पाया है। यहां वर्णित उपकरणों से परे, अतिरिक्त संसाधनों की एक भीड़ है जो जैव सूचना विज्ञान संसाधन पोर्टल जैसे कि एक्सपेसी (https://www.expasy.org) और ईएमबीएल-ईबीआई (https://www.ebi.ac.uk/services/all) में पाई जा सकती है। हालाँकि, इन रिपॉजिटरी के भीतर प्रत्येक उपकरण के लिए बारीकियों का विवरण देना इस आलेख के दायरे से परे है। यहां हम निम्नलिखित संसाधनों की सलाह देते हैं।
सबसे पहले, टीएमएचएमएम76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) ट्रांसमेम्ब्रेन डोमेन की उपस्थिति के लिए ब्याज के प्रोटीन अनुक्रमों का विश्लेषण करता है। विशेष रूप से, कई माइक्रोप्रोटीन जिन्हें कार्यात्मक रूप से अब तक विशेषता दी गई है, उनमें सिंगल-पास ट्रांसमेम्ब्रेन डोमेन होते हैं, जो झिल्ली क्षेत्रों में उनके स्थानीयकरण की सुविधा प्रदान करते हैं और आयन चैनलों, एक्सचेंजर्स और झिल्ली से जुड़े एंजाइमों के उनके प्रत्यक्ष विनियमन को सक्षम बनाता है30. दूसरा, नेशनल सेंटर फॉर बायोटेक्नोलॉजी इंफॉर्मेशन (एनसीबीआई) संरक्षित डोमेन सर्च77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) एक लोकप्रिय उपकरण है जिसका उपयोग प्रोटीन या कोडिंग न्यूक्लियोटाइड अनुक्रमों के भीतर संरक्षित डोमेन की पहचान करने के लिए किया जाता है। तीसरा, प्रोटीन परिवार (पीएफएएम) 78 डेटाबेस (http://pfam.xfam.org) प्रोटीन परिवारों और डोमेन के संरेखण और वर्गीकरण प्रदान करता है। चौथा, डब्ल्यूओएलएफ पीएसओआरटी79 (https://wolfpsort.hgc.jp/) एक उपकरण है जिसे उपकोशिकीय प्रोटीन स्थानीयकरण की भविष्यवाणी करने के लिए नियोजित किया जा सकता है। पांचवां, कॉक्सप्रेस्डबी80 एक जीन सह-अभिव्यक्ति डेटाबेस (https://coxpresdb.jp) है जो जीन कार्यों का अनुमान लगाने के लिए सह-विनियमित जीन संबंध प्रदान करता है। अंत में, सिग्नलपी 6.081 एक व्यापक रूप से उपयोग किया जाने वाला भविष्यवाणी कार्यक्रम (https://services.healthtech.dtu.dk/service.php?SignalP) है जो सिग्नल पेप्टाइड अनुक्रम की उपस्थिति को पहचानता है और दरार साइट के स्थान की भविष्यवाणी करता है।
संक्षेप में, यहां वर्णित विधियों का उपयोग यूसीएससी जीनोम ब्राउज़र पर फिलोसीएसएफ का उपयोग करके प्रोटीन-कोडिंग क्षमता के लिए ब्याज के जीनोमिक क्षेत्रों का प्रभावी ढंग से विश्लेषण करने के लिए किया जा सकता है। ये विधियां अत्यधिक सुलभ हैं और जैव सूचना विज्ञान या तुलनात्मक जीनोमिक्स में पूर्व प्रशिक्षण या विशेषज्ञता के बिना व्यक्तियों द्वारा आसानी से सीखा और कुशलतापूर्वक लागू किया जा सकता है। जैसा कि यहां विस्तार से दिखाया गया है, फिलोसीएसएफ एक शक्तिशाली उपकरण है जिसे कशेरुक, अकशेरुकी और वायरल जीनोम में प्रोटीन-कोडिंग बनाम नॉनकोडिंग जीन को अलग करने में मदद करने के लिए पहले-पास विश्लेषण के रूप में लागू किया जा सकता है, और इस दृष्टिकोण की ताकत नोट की गई कमजोरियों से भारी रूप से आगे निकल जाती है।
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
लेखकों ने घोषणा की है कि उनके पास कोई प्रतिस्पर्धी वित्तीय हित नहीं हैं।
Acknowledgments
इस काम को नेशनल इंस्टीट्यूट ऑफ हेल्थ (एचएल -141630 और एचएल -160569) और सिनसिनाटी चिल्ड्रन रिसर्च फाउंडेशन (ट्रस्टी अवार्ड) से अनुदान द्वारा समर्थित किया गया था।
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
