

Summary
여기에 설명 된 프로토콜은 사용자 친화적 인 UCSC 게놈 브라우저에서 PhyloCSF를 사용하여 마이크로 단백질 코딩 잠재력에 대한 게놈 영역을 분석하는 방법에 대한 자세한 지침을 제공합니다. 또한, 그들의 추정 기능에 대한 통찰력을 얻기 위해 확인 된 마이크로 단백질의 서열 특성을 더 조사하기 위해 몇 가지 도구와 자원이 권장됩니다.
Abstract
차세대 염기서열 분석(NGS)은 유전체학 분야를 발전시키고 수많은 동물 종 및 모델 유기체에 대한 전체 게놈 서열을 생산했습니다. 그러나 이러한 풍부한 서열 정보에도 불구하고 포괄적 인 유전자 주석 노력은 특히 작은 단백질의 경우 도전적인 것으로 입증되었습니다. 주목할 만하게, 종래의 단백질 주석 방법은 게놈 전체에 걸쳐 기하급수적으로 더 많은 수의 스퓨리어스 비코딩 sORF를 걸러내기 위해 길이가 300개 미만인 짧은 오픈 리딩 프레임(sORFs)에 의해 코딩되는 추정 단백질을 의도적으로 배제하도록 설계되었다. 그 결과, 마이크로단백질(<100개 아미노산 길이)이라고 불리는 수백 개의 기능적 작은 단백질이 비코딩 RNA로 잘못 분류되거나 완전히 간과되었다.
여기서 우리는 진화적 보존에 기초한 마이크로단백질 코딩 잠재력에 대한 게놈 영역을 쿼리하기 위해 공개적으로 이용 가능한 자유롭고 공개적으로 이용 가능한 생물정보학 도구를 활용하는 상세한 프로토콜을 제공한다. 특히, 우리는 사용자 친화적 인 캘리포니아 산타 크루즈 대학 (UCSC) 게놈 브라우저에서 계통 발생 코돈 치환 주파수 (PhyloCSF)를 사용하여 서열 보존 및 코딩 가능성을 검사하는 방법에 대한 단계별 지침을 제공합니다. 또한, 우리는 아미노산 서열 보존을 시각화하고 예측 된 도메인 구조를 포함하여 마이크로 단백질 특성을 분석하기위한 자원을 권장하기 위해 확인 된 마이크로 단백질 서열의 여러 종 정렬을 효율적으로 생성하는 단계를 자세히 설명합니다. 이러한 강력한 도구는 비정준 게놈 영역에서 추정적 마이크로단백질 코딩 서열을 확인하거나 관심있는 비코딩 전사체에서 번역 잠재력을 갖는 보존된 코딩 서열의 존재를 배제하는 데 사용될 수 있다.
Introduction
게놈에서 코딩 요소의 완전한 세트의 확인은 인간 게놈 프로젝트의 개시 이후 주요 목표였으며, 생물학적 시스템에 대한 이해와 유전 기반 질병의 병인학을 향한 핵심 목표로 남아 있습니다 1,2,3,4. NGS 기술의 발전은 척추동물, 무척추동물, 효모 및 식물5을 포함한 광범위한 수의 유기체에 대한 전체 게놈 서열의 생산으로 이어졌다. 추가적으로, 고처리량 전사 시퀀싱 방법은 세포 전사체의 복잡성을 더욱 밝혀냈고, 단백질 코딩 및 비코딩 기능 모두를 갖는 수천 개의 새로운 RNA 분자를 확인하였다6,7. 이러한 방대한 양의 서열 정보를 해독하는 것은 진행중인 과정이며, 포괄적 인 유전자 주석 노력8에 대한 과제가 남아 있습니다.
리 보솜 프로파일링9,10 및 폴리리보솜 시퀀싱 11을 포함한 번역 프로파일링 방법의 최근 개발은 수백 개의 비정규 번역 사건이 게놈 전체에 걸쳐 현재 주석이 없는 sORF에 매핑된다는 증거를 제공했으며, 마이크로단백질 또는 마이크로펩티드12,13,14,15,16이라고 불리는 작은 단백질을 생성할 가능성이 있으며, 17. 마이크로 단백질은 작은 크기 (<100 아미노산)와 고전적인 단백질 코딩 유전자 특성 8,12,18,19,20의 부족으로 인해 표준 유전자 주석 방법에 의해 간과 된 다목적 단백질의 새로운 클래스로 부상했습니다. 미세단백질 은 효모21,22, 파리 17,23,24, 포유류25,26,27,28을 포함한 거의 모든 유기체에서 기술되었으며, 발달, 대사 및 스트레스 신호 전달 19,20,29를 포함한 다양한 과정에서 중요한 역할을 하는 것으로 나타났습니다. 30,31,32,33,34. 따라서, 오랫동안 간과되어 온 기능적 작은 단백질 클래스의 추가 구성원을 위해 게놈을 계속 채굴하는 것이 필수적입니다.
마이크로 단백질의 생물학적 중요성에 대한 광범위한 인식에도 불구하고,이 부류의 유전자는 게놈 주석에서 크게 과소 대표되고 있으며, 정확한 식별은 현장에서 진전을 방해하는 지속적인 도전으로 계속되고 있습니다. 마이크로단백질 코딩 서열을 확인하는 것과 관련된 어려움을 극복하기 위해 다양한 전산 도구 및 실험 방법이 최근에 개발되었다(몇몇 포괄적인 리뷰 8,35,36,37에서 광범위하게 논의됨). 최근의 많은 미세단백질 동정 연구 38,39,40,41,42,43,44,45,46,47 은 PhyloCSF 48,49라고 불리는 알고리즘 중 하나의 사용에 크게 의존해 왔다. , 게놈의 보존된 단백질 코딩 영역과 비코딩되는 영역을 구별하기 위해 활용될 수 있는 강력한 비교 유전체학 접근법.
PhyloCSF는 단백질 코딩 유전자의 진화적 시그니처를 검출하기 위해 다종 뉴클레오티드 정렬과 계통발생 모델을 사용하는 코돈 치환 빈도(CSF)를 비교한다. 이러한 경험적 모델-기반 접근법은 단백질이 뉴클레오티드 서열보다는 아미노산 수준에서 주로 보존된다는 전제에 의존한다. 따라서, 동일한 아미노산을 인코딩하는 동의어 코돈 치환, 또는 보존된 특성(즉, 전하, 소수성, 극성)을 갖는 아미노산에 대한 코돈 치환은 긍정적으로 점수가 매겨지는 반면, 오센스 및 넌센스 치환을 포함하는 비동의어 치환은 부정적으로 점수가 매겨진다. PhyloCSF는 전체 게놈 데이터에 대해 훈련되고 표준 단백질 코딩 유전자48,49의 미세 단백질 또는 개별 엑손을 분석 할 때 필요한 전체 서열로부터 분리 된 코딩 서열 (CDS)의 짧은 부분을 채점하는 데 효과적이라는 것이 입증되었습니다.
특히, 캘리포니아 산타크루즈 대학(UCSC) 게놈 브라우저(49,50,51)에 PhyloCSF 트랙 허브의 최근 통합은 모든 배경의 조사관이 단백질 코딩 잠재력에 대한 관심 게놈 영역을 쿼리하기 위해 사용자 친화적인 인터페이스에 쉽게 액세스할 수 있게 한다. 아래에 요약된 프로토콜은 UCSC 게놈 브라우저에 PhyloCSF 트랙 허브를 로드하는 방법에 대한 자세한 지침을 제공하며, 이어서 관심 게놈 영역을 조사하여 고신뢰도 단백질 코딩 영역(또는 그 부족)을 조사합니다. 추가적으로, 양성 PhyloCSF 스코어가 관찰되는 경우에, 마이크로단백질 코딩 잠재력을 추가로 분석하고 종간 서열 보존을 예시하기 위해 확인된 아미노산 서열의 다중 종 정렬을 효율적으로 생성하기 위한 단계들이 기술된다. 마지막으로, 예측된 도메인 구조 및 추정적 마이크로단백질 기능에 대한 통찰력을 포함하여 확인된 마이크로단백질 특성을 조사하기 위해 논의에 몇 가지 추가적인 공개적으로 이용 가능한 자원 및 도구가 도입된다.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
아래에 설명 된 프로토콜은 UCSC 게놈 브라우저에서 PhyloCSF 브라우저 트랙을로드하고 탐색하는 단계를 자세히 설명합니다 (Mudge et al.49에 의해 생성됨). UCSC 게놈 브라우저에 관한 일반적인 질문은 광범위한 게놈 브라우저 사용자 안내서가 여기에서 찾을 수 있습니다 : https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. PhyloCSF 트랙 허브를 UCSC 게놈 브라우저에 로드
- 인터넷 브라우저 창을 열고 UCSC 게놈 브라우저(https://genome.ucsc.edu/)로 이동합니다.
- 도구 제목 아래에서 트랙 허브 옵션을 선택합니다.
참고: 트랙 허브 옵션은 내 데이터 탭에서도 찾을 수 있습니다. - 공용 허브 탭에서 검색 용어 상자에 PhyloCSF를 입력합니다. 공용 허브 검색 단추를 클릭합니다.
- 허브 이름 PhyloCSF에 대한 연결 버튼을 클릭하여 PhyloCSF에 연결합니다 (설명 : PhyloCSF 로 측정 된 진화 단백질 코딩 잠재력).
참고: 이 트랙 허브는 사람(hg19 및 hg38) 및 마우스(mm10 및 mm39)를 포함한 수많은 어셈블리에 로드됩니다. - 연결을 클릭 한 후 UCSC 게놈 브라우저 게이트웨이 페이지 (https://genome.ucsc.edu/cgi-bin/hgGateway)로 리디렉션 될 때까지 기다립니다.
2. 유전자 식별자를 사용하여 관심있는 유전자로 이동
- 쿼리할 종과 게놈 어셈블리를 선택합니다. 다른 종(예: 마우스)을 쿼리하려면 해당 아이콘을 클릭하여 찾아보기/ 종 선택 제목 아래에서 관심 종을 선택하거나 "종 , 일반 이름 또는 어셈블리 ID 입력"이라는 텍스트 상자에 종을 입력합니다.
참고: 어셈블리는 위치 찾기 제목 바로 아래에 나열됩니다. 전형적으로, 디폴트는 휴먼 어셈블리(예를 들어, Dec. 2009 [GRCh37/hg19])이다. - 드롭다운 메뉴를 사용하여 위치 찾기 제목 아래에서 검색할 어셈블리를 선택합니다.
- 위치/검색어 상자에 위치, 유전자 기호 또는 검색어를 입력하고 이동 을 클릭하여 게놈 브라우저에서 관심 있는 유전자로 이동합니다.
- 검색으로 인해 여러 개의 일치 항목이 발생한 경우 관심 위치를 선택해야 하는 페이지로 리디렉션될 때까지 기다립니다. 관심있는 적절한 유전자를 클릭하십시오.
3. 서열 정보를 사용하여 관심있는 게놈 영역으로 이동
- UCSC 게놈 브라우저(https://genome.ucsc.edu/)로 이동하여 도구 제목 아래에서 BLAT와 유사한 정렬 도구(BLAT)를 선택하여 특정 DNA 또는 단백질 서열을 쿼리합니다. 또는 도구 탭 위에 커서를 놓고 Blat 옵션을 선택하거나 다음 링크를 따르십시오: https://genome.ucsc.edu/cgi-bin/hgBlat.
- 드롭다운 메뉴를 사용하여 종(게놈)과 관심 어셈블리 를 선택합니다.
- 드롭다운 메뉴를 사용하여 쿼리 유형을 정의합니다.
- BLAT 검색 게놈 텍스트 상자에 관심 있는 시퀀스를 붙여넣고 제출을 클릭합니다.
- ACTIONS 제목 아래의 브라우저 링크를 클릭하여 관심있는 게놈 영역으로 이동하십시오.
4. PhyloCSF 트랙 데이터를 사용하여 보존된 sORF 식별
- PhyloCSF 영역을 긍정적으로 스코어링하기 위해 관심있는 게놈 영역을 시각적으로 스캔합니다 (그림 1).
참고: UCSC 게놈 브라우저에서 PhyloCSF 점수를 시각적으로 해석하는 방법에 대한 자세한 설명은 아래의 대표적인 결과 섹션을 참조하십시오. - 확대/축소 기능을 사용하여 관심 영역을 확대하여 시퀀스 특성을 검사하고 시작/정지 코돈을 검색합니다. 수동으로 확대하려면 Shift 키를 누른 채 마우스 버튼을 누른 채 관심 영역을 따라 드래그합니다. 또는 페이지 위쪽에 있는 확대 및 축소 단추를 사용하여 탐색합니다(1.5x, 3x, 10x 또는 기본 확대/축소 옵션 사용 가능).
참고: 확대/축소 버튼을 사용하기 전에 관심 영역이 화면 중간에 있도록 유전자의 위치를 변경해야 합니다. 이 작업을 수행하려면 이미지를 클릭하고 왼쪽 또는 오른쪽으로 드래그하여 게놈 영역을 원하는 대로 수평으로 이동하거나 페이지 상단의 이동 화살표를 사용합니다. - 뉴클레오티드(염기) 서열이 보일 때까지 확대합니다.
참고: 뉴클레오티드 서열은 +1 평활화된 PhyloCSF 점수 바로 위에 나타날 것이다. - 양성 스코어링된 PhyloCSF 영역의 시작과 끝 근처의 뉴클레오티드 서열을 육안으로 스캔하여 추정적 시작(ATG) 및 정지(TGA/TAA/TAG) 코돈을 확인한다.
참고: 관심있는 유전자가 DNA의 마이너스 가닥에 있다면, 시작 및 정지 코돈은 역보체가 될 것이다 (즉, 시작 코돈의 경우 CAT, 정지 코돈의 경우 TCA/TTA/CTA).
5. 다른 게놈에서 상동성 영역 보기
- 페이지 상단의 보기 제목 위에 마우스를 놓고 다른 게놈 입력(변환) 옵션을 클릭합니다.
- 새 게놈 제목 아래의 드롭다운 메뉴를 사용하여 관심 있는 게놈을 정의합니다.
- 새 어셈블리 제목 아래의 드롭다운 메뉴를 사용하여 원하는 게놈 어셈블리 를 선택한 다음 제출 단추를 클릭합니다.
- 브라우저가 유사성을 가진 새 어셈블리의 영역 목록을 반환하면 염색체 위치 링크를 클릭하여 관심 있는 상동 영역으로 이동합니다.
참고: 전체 염기(뉴클레오티드)의 백분율과 해당 영역에 의해 커버되는 스팬은 나열된 각 영역에 대해 정의될 것이다. 일치하는 염기의 비율이 높을수록 관심 영역에 대한 보존이 높아집니다. - 섹션 4에 설명된 것과 동일한 탐색 전략을 따라 시퀀스를 분석합니다.
6. 관심있는 미세 단백질에 대한 다종 서열 정렬 생성
- UCSC 게놈 브라우저에서 GENCODE 트랙에 관심 있는 유전자를 클릭하고(파란색 상자로 그림 1A에 표시됨) 유전자 설명 페이지로 이동합니다.
- 도구 및 데이터베이스에 대한 시퀀스 및 링크 제목에서 다른 종 FASTA를 읽는 테이블의 링크를 클릭하십시오.
- 관심있는 종과 관련된 상자를 클릭하여 선택하십시오. 제출을 클릭합니다. 페이지 하단에 나타나는 시퀀스를 FASTA 형식으로 복사하여 워드 프로세싱 문서에 붙여넣습니다.
- 두 번째 브라우저 창을 열고 유럽 생물 정보학 연구소 (EMBL-EBI) 웹 사이트 53,54 : https://www.ebi.ac.uk/Tools/msa/clustalo/ 에서 Clustal Omega 다중 시퀀스 정렬 도구52로 이동하십시오.
- 클립보드에 아직 남아 있는 시퀀스 파일을 지원되는 형식으로 시퀀스를 읽는 1단계의 상자에 붙여넣습니다. 페이지 하단으로 스크롤하여 제출을 클릭하십시오. 각 아미노산의 보존 정도를 나타내는 기호에 대해 정렬된 결과(검은색 글꼴)를 아래에서 살펴봅니다(기호는 표 1에 정의되어 있음).
참고: 정렬을 생성하는 데 몇 분 정도 걸릴 수 있습니다. - 아미노산 특성을 색상으로 보려면 서열 바로 위에 있는 색상 표시 링크를 클릭하여 해당 특성에 따라 아미노산을 채색합니다( 표 2에 정의됨).
- 시퀀스 정렬을 복사하여 워드 프로세싱 또는 슬라이드쇼 프로그램에 붙여넣어 그림 또는 그림 파일을 생성합니다(예: 그림 2).
참고: Courier와 같은 정렬을 위해 모노스페이스 글꼴을 사용하십시오. - Clustal Omega 결과 페이지에서 다른 출력을 보려면 해당 탭(예: 가이드 트리 또는 계통 발생 트리)을 클릭합니다.
- 결과 뷰어 탭을 클릭하여 다중 시퀀스 정렬 편집, 시각화 및 분석(55)을 전문으로 하는 무료 프로그램인 Jalview를 사용하여 시퀀스 정보를 보거나 MView 및 Simple Phylogeny56에 대한 직접 링크에 액세스할 수 있는 옵션을 확인하십시오.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
여기서는 검증된 마이크로단백질 미토레귤린(Mtln)을 예로 들어 보존된 sORF가 UCSC 게놈 브라우저에서 쉽게 시각화 및 분석할 수 있는 양성 PhyloCSF 점수를 생성하는 방법을 시연합니다. 미토레귤린은 이전에 비코딩 RNA (이전의 인간 유전자 ID LINC00116 및 마우스 유전자 ID 1500011K16Rik)로서 주석을 달았다. 비교 유전체학 및 서열 보존 분석 방법은 초기 발견 40,57,58,59,60,61에서 중요한 역할을했으며, 이러한 방법의 강도를 강조했습니다. 이 예에서는 마우스 GRCm38/mm10(2011년 12월) 어셈블리가 사용됩니다. 검색은 프로토콜 섹션 2에 기재된 바와 같이 유전자 식별자 (미토레귤린, Mtln) 또는 유전자 위치 (chr2:127,791,364-127,792,496)를 사용하여 수행될 수 있다. 대안적으로, 미토레귤린(도 2에 도시됨)에 대한 아미노산 서열은 BLAT 툴(프로토콜 섹션 3에 기재됨)을 사용하여 검색될 수 있다.
그림 1A에 표시된 것과 유사한 화면이 화면 상단에 표시되는 PhyloCSF 트랙 허브와 함께 나타납니다. Smoothed PhyloCSF 트랙 (각 코돈이 코딩 될 확률을 정의하는 숨겨진 Markov 모델로 매끄럽게 처리됨)은 여섯 개의 총 트랙으로 묘사되며, DNA의 플러스 가닥에 해당하는 세 개의 트랙 (녹색으로 PhyloCSF +1, +2 및 +3으로 표시됨)과 DNA의 마이너스 가닥에 해당하는 3 개의 트랙 (PhyloCSF -1로 빨간색으로 표시됨, -2 및 -3). 이들 트랙은 각 방향에서 관심있는 유전자에 대한 세 개의 잠재적 판독 프레임을 나타낸다. 브라우저 창에서 엑손은 인트론을 나타내는 얇은 파란색 수평선으로 연결된 파란색 사각형으로 표시됩니다. 인트로닉 영역의 화살촉은 유전자가 어느 방향으로 전사되는지를 나타냅니다 (따라서 PhyloCSF 점수에 초점을 맞출 가닥). 그림 1의 Mtln 예제에서 인트로닉 화살촉은 왼쪽을 가리킵니다. 따라서, Mtln 유전자는 DNA의 마이너스 가닥으로부터 전사되고, 관련 PhyloCSF 스코어는 -1, -2 및 -3 트랙(적색)에 묘사된다.
각 PhyloCSF 트랙은 선 아래에 밝은 녹색/빨간색으로 표시된 음수 점수 영역과 선 위에 짙은 녹색/빨간색으로 표시된 양의 점수 영역이 있는 얇은 검정색 선으로 표시됩니다. 소개에 설명된 바와 같이, 양의 PhyloCSF 점수는 코딩될 가능성이 있는 보존된 영역을 나타낸다. 특히 높은 서열 보존을 갖는 단백질 코딩 영역의 경우, 이들은 종종 안티센스 가닥 상에서 또한 긍정적인 점수를 받는다는 점에 유의한다; 그러나 PhyloCSF 점수는 일반적으로 올바른 가닥에서 더 높습니다. 예를 들어, 이것은 PhyloCSF-1 트랙에서 정확한 코딩 서열 스코어가 매우 높게 점수가 매겨지는 Mtln에 대해 도 1에서 알 수 있고, 안티센스 가닥(PhyloCSF+2 트랙) 또한 양의 스코어를 생성한다. 도 1A(블랙박스로 나타냄)에서 볼 수 있듯이, Mtln의 첫 번째 엑손에는 PhyloCSF-1 트랙에서 매우 높은 점수를 받는 영역이 있는데, 이는 이것이 코딩 영역에 해당할 수 있음을 시사한다. 이 영역을 더 자세히 조사하려면 영역을 확대하고 확대하는 것이 도움이 됩니다(그림 1B). 도 1C,D에 도시된 바와 같이, Mtln의 제1 엑손에서 양성 스코어링 영역은 시작 코돈에 걸쳐 직접 시작하고(도 1C), 정지 코돈(도 1D)에서 종결되며, 이는 ORF가 고도로 보존되고 있음을 나타내고 ORF를 코딩한다는 것을 강하게 시사한다. Mtlen이 DNA의 마이너스 가닥 상에 있는 바와 같이, 시작 및 정지 코돈은 코돈의 역방향 보체로서 도시된다(즉, ATG 개시 코돈은 CAT[도 1C]로서 도시되고, TGA 정지 코돈은 TCA로서 도시된다[도 1D]).
PhyloCSF를 사용하여 마이크로단백질 코딩 잠재력을 갖는 보존된 영역을 검색하는 것 외에도, 이 기술은 보존된 ORF의 존재를 배제하기 위해 추정적 비코딩 RNA의 최초 통과 분석으로서 적용될 수 있으며, 따라서 비코딩 주석에 대한 지원을 제공한다. 예를 들어, PhyloCSF를 이용한 잘 특성화된 lncRNA HOTAIR 62,63의 분석은 6개 트랙 모두에 걸쳐 전체 유전자에 걸쳐 음성 스코어를 보여주며(도 3), 서열 보존의 결여를 강하게 나타내고 HOTAIR가 비코딩 RNA로서 정확하게 주석을 달았다는 지지를 제공한다.
도 1에서 명확하게 볼 수 있는 바와 같이, 미토레귤린에 대한 전체 코딩 ORF는 단일 엑손 내에 위치하여, 단일의 중단되지 않고, 긍정적인 스코어링 영역을 갖는 PhyloCSF에 의한 간단하고 간단한 판독을 생성한다. 그러나 PhyloCSF 트랙 허브 데이터가 항상 명확하고 해석하기 쉬운 것은 아닙니다. 예를 들어, 마우스 1810058I24Rik 유전자 47,64,65에 의해 코딩되는 미톨람 반/Stmp1/Mm47 마이크로단백질은 3개의 엑손에 걸쳐 있는 보존된 ORF를 묘사하고(그림 4A), 양의 PhyloCSF 점수는 엑손 1의 +2 트랙(그림 4B)에서 엑손 2의 +3 트랙(그림 4C)으로 점프한 다음 엑손 3의 +2 트랙으로 다시 점프합니다(그림 4D ). 언뜻보기에는 혼란스러워 보이지만 설명은 매우 간단합니다. PhyloCSF는 각 유전자에 대한 특정 엑손/인트론 구조를 고려하지 않고 게놈 영역의 여섯 개의 잠재적 판독 프레임(DNA의 플러스 가닥에 세 개, 마이너스 가닥에 세 개)을 채점합니다. 따라서, 인트로닉 서열 정보를 리딩 프레임의 3-뉴클레오티드 주기성으로 유지한다. 따라서, 인트론이 세 개(즉, 세 개의 뉴클레오티드/코돈)로 나눌 수 없는 다수의 뉴클레오티드를 포함한다면, PhyloCSF 판독 프레임은 한 트랙에서 다른 트랙으로 점프할 것이다.
마지막으로, PhyloCSF는 또한 단일 RNA 분자 내에서 다수의 별개의 코딩 ORF를 확인하는데 효과적으로 사용될 수 있다. 예를 들어, MIEF1 마이크로단백질(MIEF1-MP)은 미토콘드리아 신장 인자 1(MIEF1)66의 5' UTR 내에서 코딩된다(도 5). MIEF1 게놈 영역이 PhyloCSF에 의해 분석될 때, MIEF1-MP에 상응하는 이산 양성 PhyloCSF 스코어(도 5C)는 MIEF1에 대한 주 CDS의 상류에서 용이하게 관찰될 수 있다(도 5B). MIEF1 및 그의 관련 미세단백질(MIEF1-MP)에 대한 추가 논의는 본 문서에 요약된 방법 및 프로토콜의 강점과 약점에 대한 요약과 함께 아래의 논의에서 제공된다.

도 1: 미토레귤린(Mtln) 유전자의 PhyloCSF 분석은 검증된 마이크로단백질에 상응하는 고서열 보존의 영역을 나타낸다. (A) UCSC 게놈 브라우저 및 PhyloCSF 트랙의 스크린 샷 은 Mtlen 에 두 개의 엑손과 단일 인트론이 포함되어 있음을 보여줍니다. 인트론 내의 화살촉은 왼쪽을 가리키며, 이는 Mtln 유전자가 DNA의 마이너스 가닥으로부터 전사되었음을 나타내며, 따라서 관련 PhyloCSF 점수는 -1, -2 및 -3 트랙(빨간색)에 표시됩니다. 완전한 미토레귤린 코딩 서열은 엑손 1 내에 포함되어 있으며 PhyloCSF-1 트랙(B)에서 높은 점수를 받는다. 보존된 시작 코돈은 녹색 박스(CAT, 역보체 ATG)로 강조되는 PhyloCSF-1 트랙(C)에서 양성 스코어링 영역의 시작에서 명확하게 관찰될 수 있다. 추가적으로, 보존된 정지 코돈 (TCA, 역보체 TGA)은 패널 (D)에 적색 박스로 표시되며, 이는 양성 스코어링 PhyloCSF 영역의 끝과 정렬된다. Mtln 유전자에 대한 상세한 정보는 파란색 상자 내의 Mtln 유전자 식별자를 클릭함으로써 찾을 수 있다(패널 A에 표시됨). 참고로, 고도로 보존된 단백질-코딩 영역은 종종 안티센스 가닥에서 긍정적으로 점수가 매겨진다( Mtln에 대한 PhyloCSF +2 트랙에서 볼 수 있다). 그러나, PhyloCSF 점수는 전형적으로 정확한 가닥에서 더 높다(이 예에서는 PhyloCSF-1 트랙). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 2: Clustal Omega 프로그램을 사용하여 생성된 마이크로단백질 미토레귤린의 다중 종 서열 정렬. 표시된 여덟 종에 대한 미토레귤린 아미노산 서열을 프로토콜 섹션 6에 상세히 설명된 대로 추출하고 Clustal Omega 다중 서열 정렬 도구와 정렬하였다. 아미노산의 특성은 색상 (적색, 소형/소수성; 청색, 산성, 자홍색, 염기성; 녹색, 히드록실/술프히드릴/아민)( 표 2에 추가로 정의됨)으로 표시된다. 아미노산 아래의 기호는 보존 정도(별표, 완전 보존된 잔기; 결장, 강하게 유사한 성질을 갖는 아미노산; 기간, 약하게 유사한 성질의 그룹들 사이의 보존)를 나타낸다( 표 1에 광범위하게 상세히 설명됨). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
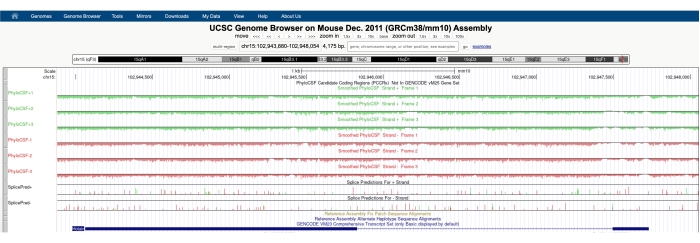
그림 3: 검증된 긴 비코딩 RNA Hotair에 대한 PhyloCSF 트랙의 스크린샷은 게놈 유전자좌 전체에 걸쳐 서열 보존의 부족을 보여준다. Hotair 의 인트로닉 영역에있는 화살촉은 왼쪽을 가리키고 있으며, 이는 lncRNA가 DNA의 음의 가닥에서 전사되었음을 나타내며, 따라서 PhyloCSF -1, -2 및 -3 트랙이 분석의 초점이되어야합니다. PhyloCSF 점수는 전체 유전자 (여섯 트랙 모두에 대해)에 걸쳐 음성이며, 이는 서열 보존의 부족을 나타내며, 이는 비코딩 RNA로서의 적절한 주석을 뒷받침합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 4: 미세단백질 미톨람반/Stmp1/Mm47을 인코딩하는 마우스 1810058I24Rik 유전자의 PhyloCSF 분석. (A) 마우스 1810058I24Rik 유전자는 세 개의 엑손으로 구성되어 있으며, 인트로닉 영역의 화살촉이 오른쪽을 가리키며, 이는 DNA의 플러스 가닥에 전사되어 있으므로 PhyloCSF +1, +2 및 +3 트랙을 분석해야 함을 나타냅니다. 보존된 마이크로단백질 코딩 서열은 엑손 1(B)에서 시작하여, 엑손 2(C)를 통해 판독하고, 엑손 3(D)로 끝나는 3개의 엑손 모두에 걸쳐 있다. 양의 PhyloCSF 점수는 엑손 1의 +2 트랙, 엑손 2의 +3 트랙 및 엑손 1의 +2 트랙에서 발견됩니다. 양성 점수가 한 트랙에서 다른 트랙으로 이동하는 이유는 PhyloCSF가 유전자의 엑손 / 인트론 구조와 독립적으로 DNA 서열의 여섯 가지 잠재적 인 판독 프레임을 분석하기 때문입니다. 따라서, 세 개(세 개의 뉴클레오티드/코돈)로 나눌 수 없는 다수의 뉴클레오티드를 포함하는 인트론은 판독 프레임에서 다른 트랙으로 이동을 야기할 것이다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

도 5: PhyloCSF를 사용한 Mief1 게놈 유전자좌의 분석은 공유 RNA 상의 주요 Mief1 CDS와 무관하게 5' UTR에서 단백질 코딩 전위를 갖는 영역을 동정한다. 이러한 보존된 상류 ORF(uORF)는 Mief1-MP라는 이름의 마이크로단백질을 인코딩하는 것으로 나타났다. (A) Mief1 게놈 유전자좌의 개요. 인트론의 화살촉은 오른쪽을 가리키며, 이는 Mief1이 DNA의 플러스 가닥에서 전사되었음을 나타냅니다 (코딩 잠재력을 결정하기 위해 PhyloCSF +1, +2 및 +3 트랙에 중점을 둡니다). 주요 Mief1 CDS는 463 아미노산 단백질을 인코딩하며 패널 (B)에 표시되어 있습니다. 그러나, Mief1-MP (C)라고 불리는 독특한 70개 아미노산 마이크로단백질을 인코딩하는 Mief1의 5' UTR 내에 뚜렷한 보존된 상류 ORF가 또한 존재한다. 패널 C에서 볼 수 있듯이, Mief1-MP는 Mief1 5' UTR 내에서 자체 보존된 시작 및 정지 코돈을 가지며, ORF는 PhyloCSF +1 트랙에서 매우 높은 점수를 받아 기능적 미세단백질을 인코딩한다는 강력한 증거를 제공합니다. 약어 : ORF = 오픈 리딩 프레임; uORF = 업스트림 ORF; UTR = 번역되지 않은 영역; CDS = 코딩 시퀀스. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
| 상징 | 아미노산 보존 수준 | 그룹화된 아미노산 |
| 별표(*) | 완전히 보존된 잔여물 | 적용 불가(단일, 완전 보존된 잔류물) |
| 결장 (:) | 매우 유사한 특성을 가진 그룹 | 스타; NEQK; NHQK; NDEQ; QHRK; MILV; 밀프; 하이; 증권 시세 표시기 |
| 기간(.) | 약하게 유사한 속성을 가진 그룹 | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| 공간(기호 없음) | 유사성 없음 | 해당 사항 없음(유사성 없음) |
표 1: Clustal Omega에 의해 생성된 다중 시퀀스 정렬에 대한 합의 기호의 정의. 도 2 에 도시된 다중 종 서열 정렬은 Clustal Omega52를 사용하여 생성되었다. 약어: 세린 (S), 트레오닌 (T), 알라닌 (A), 아스파라긴 (N), 글루탐산 (E), 글루타민 (Q), 리신 (K), 아스파르트산 (D), 아르기닌 (R), 메티오닌 (M), 이소류신 (I), 류신 (L), 페닐알라닌 (F), 히스티딘 (H), 티로신 (Y), 트립토판 (W), 시스테인 (C), 발린 (V), 글리신 (G), 프롤린 (P).
| 글꼴 색상 | 재산 | 아미노산 잔기 [약어] |
| 빨강 | 작고 소수성 | 알라닌 [A], 발린 [V], 페닐알라닌 [F], 프롤린 [P], 메티오닌 [M], 이소류신 [I], 류신 [L], 트립토판 [W] |
| 파랑 | 산성의 | 아스파르트산 [D], 글루탐산 [E] |
| 자홍색 | 기초의 | 아르기닌 [R], 라이신 [K] |
| 녹색 | 히드록슬, 설프히드릴, 아민, +G | 세린 [S], 트레오닌 [T], 티로신 [Y], 히스티딘 [H], 시스테인 [C], 아스파라긴 [N], 글리신 [G], 글루타민 [Q] |
표 2: 도 2에 묘사된 아미노산의 특성. Clustal Omega52 는 도 2에 도시된 다중 서열 정렬을 생성하는데 사용되었다.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
여기에 제시된 프로토콜은 사용자 친화적 인 UCSC 게놈 브라우저 48,49,50,51에서 PhyloCSF를 사용하여 마이크로 단백질 코딩 잠재력에 대한 관심있는 게놈 영역을 조사하는 방법에 대한 자세한 지침을 제공합니다. 위에서 설명한 바와 같이, PhyloCSF는 계통발생 모델과 코돈 치환 빈도를 통합하여 단백질 코딩 유전자48,49의 전형적인 진화적 시그니처를 확인하는 강력한 비교 유전체학 알고리즘이다. PhyloCSF는 이전에 비코딩으로 주석이 달린 게놈 영역에서 기능적 마이크로단백질을 확인하기 위해 널리 사용되어 왔으며 38,39,40,41,42,43,44,45,46,47 , 그리고 이 접근법은 13개의 아미노산만큼 작은 마이크로단백질과 같은 짧은 서열에 대한 다른 비교 유전체학 방법들 및 35,48,49개의 정준 단백질의 작은 엑손들에 대해 능가하는 것으로 나타났다. 특히, 진화적 보존을 통해 기능적 단백질 코딩 서열을 확인하는 강력한 방법으로서의 PhyloCSF의 유용성은 척추동물 및 무척추동물 종의 한계를 넘어 최근에는 SARS-CoV-2 게놈67의 단백질 코딩 능력을 성공적으로 조사하기 위해 바이러스 게놈에 적용되고 있다.
주석이 달린 비코딩 RNA 내에서 추정적 코딩 서열을 확인하는 것 외에도, PhyloCSF의 장점은 5' 업스트림 및 3' 다운스트림 ORF(각각 uORFs 및 dORFs)를 모두 포함하는 정식 단백질 코딩 유전자의 주석이 달린 비번역 영역(UTR) 내에서 ORF에 의해 코딩되는 보존된 마이크로단백질을 안정적으로 검출할 수 있다는 것이다.8,19,66,68 . 예를 들어, MIEF1 마이크로단백질 (MIEF1-MP)은 미토콘드리아 신장 인자 1 (MIEF1)66의 5' UTR에서 코딩된다. MIEF1-MP의 경우에, MIEF1-MP에 상응하는 이산 양성 PhyloCSF 스코어가 MIEF1을 인코딩하는 ORF의 상류에서 관찰된다(도 5). 일부 uORF 인코딩된 마이크로단백질은 그들의 공유 mRNA, (예를 들어 MIEF1-MP 및 MIEF1) 상의 다운스트림 정준 단백질과 직접 상호작용하는 반면, 다른 것들은 주 CDS66,68에 의해 코딩되는 단백질과 독립적으로 기능한다. 따라서, uORF로 코딩된 마이크로단백질을 특성화할 때, 이들이 그들의 하류 단백질 생성물의 직접적인 조절을 통해 기능한다고 가정해서는 안 된다.
PhyloCSF는 보존된 마이크로단백질 코딩 서열의 확인을 위한 도구로서 많은 명확한 강점을 가지고 있지만, 이 방법의 몇 가지 한계를 인식하는 것이 중요하다. 첫째, 서열 보존은 게놈 영역이 기능적 선택을 거쳐 코딩되고 있음을 강력하게 시사하는 반면, 강력한 보존의 부족과 결과적으로 부정적인 PhyloCSF 점수가 주어진 서열에 대한 코딩 잠재력을 결정적으로 배제하지는 않는다. 즉, PhyloCSF에만 전적으로 의존하는 것은 강력하게 보존되지 않지만 기능적 미세 단백질을 생산하는 번역 된 ORF를 감독 할 수 있습니다. 특히, 보존성 또는 음성 보존 점수가 낮은 게놈 영역은 종 특이적 코딩 영역 또는 서열 발산 또는 de novo 유전자 탄생 46,69,70,71,72,73,74 를 통한 진화적 "젊은" 유전자의 유전자에 상응할 수 있다. 예를 들어, 이전에 인간 비코딩 RNA LINC00467로 생각되었던 것에 의해 코딩되는 미세단백질 ASAP는 아미노산 서열이 더 높은 포유동물(75)에서만 보존되기 때문에 PhyloCSF에 의해 긍정적으로 점수가 매겨지지 않는다. 추가적으로, 최근의 연구들은 양성 PhyloCSF 스코어68,72를 생성하지 않는 인터제닉 lncRNA RP3-527G5.1에 의해 코딩되는 것을 포함하는 몇몇 인간 특이적 마이크로단백질을 확인했다. 이와 관련하여, 양성 PhyloCSF 점수의 부재는 비코딩 영역의 증거로 해석될 수 없으며, 주의해서 해석되어야 한다.
PhyloCSF를 사용할 때 명심해야 할 두 번째 고려 사항은 긍정적 인 점수가 기능적 선택 및 단백질 코딩 능력을 매우 암시하지만 이러한 증거 라인은 단독으로 설 수 없으며 실험적으로 검증되어야한다는 것입니다. 안정한 마이크로단백질 발현에 대한 지지증거를 생성하는데 사용될 수 있는 방법의 예는 관심있는 마이크로단백질 서열에 대해 상승된 항체를 이용한 질량 분광법 또는 웨스턴 블롯팅에 의한 추정 단백질의 검출을 포함한다. 대안적으로, 최적의 항원성을 위한 서열 선택의 부족으로 인해 마이크로단백질에 대한 신뢰할 수 있는 항체를 생성하는 것이 어려울 수 있기 때문에, CRISPR/Cas9 및 상동성-지향 복구(HDR) 경로를 사용하여 추정성 마이크로단백질 서열과 함께 프레임내의 내인성 유전자좌에 에피토프 태그를 도입하여, 고친화성 항체를 사용하여 관심있는 단백질의 검출을 용이하게 할 수 있다 (예를 들어, 플래그, HA, V5, Myc)18. PhyloCSF의 최종 한계는 현재 호모 사피엔스 (인간 hg19, hg38), 무스 근육 (마우스 mm10, mm39), 갈 루스 갈루스 (닭, galGal4, galGal6), 초파리 멜라노가스터 (초파리 , dm6), Caenorhabditis elegans 등 일반적으로 사용되는 게놈 어셈블리의 많은 부분에 통합되어 있지만 (선충류, ce11) 및 SARS-CoV-2 (wuhCor1)는 현재 UCSC 게놈 브라우저에서 직접 쿼리 할 수없는 많은 종들이 여전히 있습니다.
확인된 마이크로단백질 내에서 보존된 도메인 또는 서열 특성의 확인은 그들의 기능적 관련성에 대한 신뢰를 증가시키고 그들의 추정 기능에 대한 약간의 통찰력을 제공하는 데 도움이 될 수 있다. 여기서 우리는 그러한 통찰력을 얻기 위해 확인 된 미세 단백질 아미노산 서열을 더 자세히 분석하는 데 사용할 수있는 특정 도구 및 자원에 대한 권장 사항을 제공합니다. 아래에 열거 된 특정 도구 (및 재료 표에 요약되어 있음)는 대중에게 자유롭게 제공되며 마이크로 단백질 연구18,38,39,40,41,47에서 특히 사용자 친화적이고 강력한 것으로 나타났습니다. 여기에 설명 된 도구 외에도 Expasy (https://www.expasy.org) 및 EMBL-EBI (https://www.ebi.ac.uk/services/all)와 같은 생물 정보학 자원 포털에서 찾을 수있는 많은 추가 리소스가 있습니다. 그러나 이러한 저장소 내의 각 도구에 대한 세부 정보를 자세히 설명하는 것은 이 문서의 범위를 벗어납니다. 여기서는 다음 리소스를 사용하는 것이 좋습니다.
먼저, TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0)은 막횡단 도메인의 존재에 대해 관심있는 단백질 서열을 분석한다. 특히, 지금까지 기능적으로 특성화되어 온 다수의 미세단백질은 단일 통과 막횡단 도메인을 함유하며, 이는 막 영역으로의 국소화를 용이하게 하고 이온 채널, 교환기 및 막 관련 효소(30)의 직접적인 조절을 가능하게 한다. 둘째, NCBI (National Center for Biotechnology Information) 보존 도메인 검색77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)은 단백질 또는 코딩 뉴클레오티드 서열 내에서 보존 된 도메인을 확인하는 데 사용되는 인기있는 도구입니다. 셋째, 단백질 패밀리 (Pfam)78 데이터베이스 (http://pfam.xfam.org)는 단백질 패밀리 및 도메인의 정렬 및 분류를 제공한다. 넷째, WoLF PSORT79 (https://wolfpsort.hgc.jp/)는 아세포 단백질 국소화를 예측하는데 사용될 수 있는 도구이다. 다섯째, COXPRESdB80 은 유전자 기능을 추정하기 위해 공동-조절된 유전자 관계를 제공하는 유전자 공동발현 데이터베이스(https://coxpresdb.jp)이다. 마지막으로, SignalP 6.081 은 신호 펩티드 서열의 존재를 인식하고 절단 부위의 위치를 예측하는 널리 사용되는 예측 프로그램(https://services.healthtech.dtu.dk/service.php?SignalP)이다.
요약하면, 여기에 기술된 방법은 UCSC 게놈 브라우저 상의 PhyloCSF를 사용하여 단백질 코딩 잠재력에 대한 관심 게놈 영역을 효과적으로 분석하는데 사용될 수 있다. 이러한 방법은 접근성이 뛰어나며 생물 정보학 또는 비교 유전체학에 대한 사전 교육이나 전문 지식없이 개인이 쉽게 배우고 효율적으로 적용 할 수 있습니다. 여기에서 자세히 설명했듯이, PhyloCSF는 척추동물, 무척추동물 및 바이러스 게놈에서 단백질 코딩 유전자와 비코딩 유전자를 구별하는 데 도움이 되는 첫 번째 통과 분석으로 적용할 수 있는 강력한 도구이며, 이 접근법의 강점은 주목받는 약점보다 훨씬 큽니다.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
저자들은 경쟁적인 재정적 이익이 없다고 선언합니다.
Acknowledgments
이 연구는 국립 보건원 (HL-141630 및 HL-160569)과 신시내티 아동 연구 재단 (수탁자 상)의 보조금으로 지원되었습니다.
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
