

Summary
Protokollen som er beskrevet her gir detaljerte instruksjoner om hvordan du analyserer genomiske interesseområder for mikroproteinkodingspotensial ved hjelp av PhyloCSF på den brukervennlige UCSC Genome Browser. I tillegg anbefales flere verktøy og ressurser å undersøke sekvensegenskapene til identifiserte mikroproteiner ytterligere for å få innsikt i deres putative funksjoner.
Abstract
Neste generasjons sekvensering (NGS) har drevet genomikkfeltet fremover og produsert hele genomsekvenser for mange dyrearter og modellorganismer. Til tross for denne rikdommen av sekvensinformasjon har imidlertid omfattende genmerknadsarbeid vist seg utfordrende, spesielt for små proteiner. Spesielt ble konvensjonelle proteinmerknadsmetoder designet for med vilje å utelukke putative proteiner kodet av korte åpne leserammer (sORFer) mindre enn 300 nukleotider i lengden for å filtrere ut det eksponentielt høyere antallet falske ikke-kodingssorfer i hele genomet. Som et resultat har hundrevis av funksjonelle små proteiner kalt mikroproteiner (<100 aminosyrer i lengden) blitt feilaktig klassifisert som ikke-koding av RNAer eller oversett helt.
Her tilbyr vi en detaljert protokoll for å utnytte gratis, offentlig tilgjengelige bioinformatiske verktøy for å spørre genomiske regioner for mikroproteinkodingspotensial basert på evolusjonær bevaring. Spesielt gir vi trinnvise instruksjoner om hvordan du undersøker sekvensbevaring og kodingspotensial ved hjelp av Phylogenetic Codon Substitution Frequencies (PhyloCSF) på den brukervennlige University of California Santa Cruz (UCSC) Genome Browser. I tillegg beskriver vi trinn for effektivt å generere flere arter justeringer av identifiserte mikroproteinsekvenser for å visualisere aminosyresekvensbevaring og anbefale ressurser for å analysere mikroproteinegenskaper, inkludert anslåtte domenestrukturer. Disse kraftige verktøyene kan brukes til å identifisere putative mikroproteinkodingssekvenser i ikke-kanoniske genomiske regioner eller for å utelukke tilstedeværelsen av en bevart kodingssekvens med translasjonspotensial i en ikke-koding transkripsjon av interesse.
Introduction
Identifiseringen av det komplette settet med kodeelementer i genomet har vært et hovedmål siden oppstarten av Human Genome Project, og er fortsatt et sentralt mål for forståelsen av biologiske systemer og etiologien til genetisk baserte sykdommer 1,2,3,4. Fremskritt innen NGS-teknikker har ført til produksjon av hele genomsekvenser for et omfattende antall organismer, inkludert vertebrater, hvirvelløse dyr, gjær og planter5. I tillegg har transkripsjonsmetoder med høy gjennomstrømning ytterligere avslørt kompleksiteten i det cellulære transkripsjonsomet, og identifisert tusenvis av nye RNA-molekyler med både proteinkoding ogikke-kodingsfunksjoner 6,7. Dekoding av denne enorme mengden sekvensinformasjon er en pågående prosess, og utfordringene forblir med omfattende genmerknadsarbeid8.
Den nylige utviklingen av translasjonsprofileringsmetoder, inkludert ribosomprofilering 9,10 og poly-ribosomsekvensering11, har gitt bevis som indikerer at hundrevis av ikke-kanoniske oversettelseshendelser kartlegger til for tiden uannotert sORFer gjennom hele genomet, med potensial til å generere små proteiner kalt mikroproteiner eller mikropeptider 12,13,14,15,16, 17. Mikroproteiner har dukket opp som en ny klasse allsidige proteiner som tidligere ble oversett av standard genmerknadsmetoder på grunn av deres lille størrelse (<100 aminosyrer) og mangel på klassiske proteinkodingsgenegenskaper 8,12,18,19,20. Mikroproteiner har blitt beskrevet i nesten alle organismer, inkludert gjær21,22, flyr 17,23,24, og pattedyr 25,26,27,28, og har vist seg å spille kritiske roller i ulike prosesser, inkludert utvikling, metabolisme ogstresssignalering 19,20,29, 30,31,32,33,34. Dermed er det viktig å fortsette å utvinne genomet for flere medlemmer av denne lenge oversett klassen av funksjonelle små proteiner.
Til tross for den utbredte anerkjennelsen av mikroproteiners biologiske betydning, forblir denne klassen av gener svært underrepresentert i genommerknader, og deres nøyaktige identifikasjon fortsetter å være en pågående utfordring som har hindret fremgang i feltet. Ulike beregningsverktøy og eksperimentelle metoder har nylig blitt utviklet for å overvinne vanskelighetene forbundet med å identifisere mikroproteinkodingssekvenser (diskutert mye i flere omfattende vurderinger 8,35,36,37). Mange nylige mikroproteinidentifikasjonsstudier 38,39,40,41,42,43,44,45,46,47 har stolt sterkt på bruken av en slik algoritme kalt PhyloCSF48,49 , en kraftig komparativ genomisk tilnærming som kan utnyttes til å skille konserverte proteinkodingsregioner av genomet fra de som ikke er koding.
PhyloCSF sammenligner codon substitusjon frekvenser (CSF) ved hjelp av multi-arter nukleotid justeringer og fylogenetiske modeller for å oppdage evolusjonære signaturer av protein-koding gener. Denne empiriske modellbaserte tilnærmingen er avhengig av forutsetningen om at proteiner primært bevares på aminosyrenivå i stedet for nukleotidsekvensen. Derfor skåres synonyme codon-substitusjoner, som koder den samme aminosyren, eller kodonerstatninger til aminosyrer med konserverte egenskaper (dvs. ladning, hydrofobi, polaritet) positivt, mens ikke-synonyme substitusjoner, inkludert missense og tullerstatninger, scorer negativt. PhyloCSF er opplært på fullgenomdata og har vist seg å være effektiv i å score korte deler av en kodesekvens (CDS) isolert fra hele sekvensen, noe som er nødvendig når du analyserer mikroproteiner eller individuelle eksoner av standard proteinkodingsgener48,49.
Spesielt gjør den nylige integrasjonen av PhyloCSF-sporknutepunktene i University of California Santa Cruz (UCSC) Genome Browser 49,50,51 det mulig for etterforskere av alle bakgrunner å enkelt få tilgang til et brukervennlig grensesnitt for å spørre genomiske interesseregioner for proteinkodingspotensial. Protokollen som er skissert nedenfor gir detaljert instruksjon om hvordan du laster inn PhyloCSF-sporhubene på UCSC Genome Browser og deretter forhører genomiske interesseregioner for å sondere for proteinkodingsregioner med høy tillit (eller mangelen på det). I tillegg, i tilfelle der en positiv PhyloCSF-poengsum observeres, avgrenses trinn for å analysere mikroproteinkodingspotensialet ytterligere og effektivt generere flere artsjusteringer av de identifiserte aminosyresekvensene for å illustrere sekvensbevaring på tvers av arter. Til slutt introduseres flere ekstra offentlig tilgjengelige ressurser og verktøy i diskusjonen for å kartlegge identifiserte mikroproteinegenskaper, inkludert anslåtte domenestrukturer og innsikt i putativ mikroproteinfunksjon.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Protokollen som er skissert nedenfor beskriver trinn for å laste og navigere i PhyloCSF-nettlesersporene på UCSC Genome Browser (generert av Mudge et al.49). For generelle spørsmål angående UCSC Genome Browser, finner du en omfattende Genom nettleser brukerhåndbok her: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Lasting av PhyloCSF Track Hub til UCSC Genome Browser
- Åpne et nettleservindu og naviger til UCSC Genome Browser (https://genome.ucsc.edu/).
- Velg Alternativet Spor huber under overskriften Våre verktøy.
MERK: Spor huber-alternativet finner du også under Mine data-fanen . - I kategorien Offentlige huber skriver du inn PhyloCSF i Boksen Søkeord . Klikk på Søk i offentlige huber-knappen .
- Koble til PhyloCSF ved å klikke på Koble til-knappen for Hub Name PhyloCSF (Beskrivelse: Evolusjonært proteinkodingspotensial målt ved PhyloCSF).
MERK: Denne sporhuben lastes inn i mange samlinger, inkludert menneskelig (hg19 og hg38) og mus (mm10 og mm39). - Etter å ha klikket på koble til, vent med å bli omdirigert til UCSC Genome Browser Gateway-siden (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Navigere til gener av interesse ved hjelp av genidentifikatorer
- Velg arten og genomsamlingen som skal spørres. Hvis du vil spørre etter en annen art (f.eks. mus), velger du artene av interesse under overskriften Bla gjennom / velg arter ved å klikke på det aktuelle ikonet, eller skriv inn arten i tekstboksen som sier Skriv inn arter, vanlig navn eller monterings-ID.
MERK: Monteringen er oppført direkte under overskriften Finn posisjon . Standarden er vanligvis human assembly (f.eks. des. 2009 [GRCh37/hg19]). - Velg samlingen du vil søke i, under overskriften Søk etter posisjon ved hjelp av rullegardinmenyen.
- Skriv inn posisjonen, gensymbolet eller søkeordene i Posisjon/søkeord-boksen , og klikk på Gå til for å navigere til et gen av interesse i Genom-nettleseren.
- Hvis søket resulterte i flere treff, venter du på å bli omdirigert til en side som krever valg av en interesseposisjon. Klikk på det aktuelle genet av interesse.
3. Navigere til genomiske interesseområder ved hjelp av sekvensinformasjon
- Naviger til UCSC Genome Browser (https://genome.ucsc.edu/) og velg BLAST-Like Alignment Tool (BLAT) under overskriften Våre verktøy for å spørre en bestemt DNA- eller proteinsekvens. Alternativt kan du holde markøren over Verktøy-fanen og velge Blat-alternativet eller følge denne lenken: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Velg arten (Genome) og Interessesamlingen ved hjelp av rullegardinmenyene.
- Definer spørringstypen ved hjelp av rullegardinmenyen.
- Lim inn sekvensen av interesse i BLAT Search Genome-tekstboksen , og klikk Send.
- Klikk på nettleserkoblingen under ACTIONS-overskriften for å navigere til det genomiske interesseområdet.
4. Identifisere bevarte sORFer ved hjelp av PhyloCSF Track Data
- Skann det genomiske interesseområdet visuelt for å få en positiv skåring av PhyloCSF-regioner (figur 1).
MERK: For en detaljert forklaring av hvordan du visuelt tolker PhyloCSF-score på UCSC Genome Browser, se avsnittet om representative resultater nedenfor. - Bruk zoomfunksjonen til å forstørre områder av interesse for å undersøke sekvensegenskaper og søke etter start/stopp-codons. Hvis du vil zoome inn manuelt, holder du nede SKIFT-tasten og klikker og holder nede museknappen mens du drar langs interesseområdet. Alternativt kan du bruke zoom inn- og zoome ut-knappene øverst på siden til å navigere (alternativer for 1,5x, 3x, 10x eller base zoom er tilgjengelige).
MERK: Før du bruker zoom inn / zoom ut-knappene , er det nødvendig å omplassere genet slik at interesseområdet er midt på skjermen. For å utføre denne handlingen, klikk på bildet og dra det til venstre eller høyre for å flytte det genomiske området horisontalt etter ønske eller bruk flyttepilene øverst på siden. - Zoom inn til nukleotidsekvensen (basissekvensen) er synlig.
MERK: Nukleotidsekvensen vises rett over +1 Utjevnet PhyloCSF-poengsum. - Skann nukleotidsekvensen visuelt nær begynnelsen og slutten av de positivt skårende PhyloCSF-områdene for å identifisere putative start-kodoner (ATG) og stoppkodinger (TGA/TAA/TAG).
MERK: Hvis genet av interesse er på minus dna-strengen, vil start- og stoppkokonene være omvendt komplement (dvs. CAT for startkodon og TCA / TTA / CTA for stoppkodon).
5. Visning av homologe regioner i andre genomer
- Hold musen over Visningsoverskriften øverst på siden, og klikk alternativet I andre genomer (konverter).
- Definer interessegenomet ved hjelp av rullegardinmenyen under overskriften Nytt genom .
- Velg den genomiske samlingen av interesse ved hjelp av rullegardinmenyen under overskriften Ny samling , og klikk deretter Send-knappen .
- Når nettleseren returnerer en liste over regioner i den nye samlingen med likhet, klikker du på kromosomposisjonskoblingen for å navigere til den homologe interesseområdet.
MERK: Prosentandelen av totale baser (nukleotider) og spennet som dekkes av regionen vil bli definert for hver region som er oppført. Jo høyere prosentandel av matchende baser, jo høyere er bevaringen for interesseområdet. - Følg de samme navigasjonsstrategiene som er beskrevet i avsnitt 4 for å analysere sekvensen.
6. Generering av sekvensjusteringer av flere arter for mikroproteiner av interesse
- Klikk på genet av interesse i GENCODE-sporet på UCSC Genome Browser (angitt i figur 1A med en blå boks) for å navigere til genbeskrivelsessiden.
- Under overskriften Sekvens og koblinger til verktøy og databaser klikker du koblingen i tabellen som leser Andre arter FASTA.
- Klikk på boksene som er knyttet til arten av interesse for å velge dem. Klikk på Send. Kopier og lim inn sekvensene som vises nederst på siden i FASTA-format, i et tekstbehandlingsdokument.
- Åpne et nytt nettleservindu og naviger til Clustal Omega Multiple Sequence Alignment tool52 på nettstedet til European Bioinformatics Institute (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Lim inn sekvensfilene som fremdeles er på utklippstavlen, i boksen i TRINN 1 som leser sekvenser i et hvilket som helst format som støttes. Bla til bunnen av siden og klikk på Send. Se under de justerte resultatene (i svart skrift) for symboler som angir graden av bevaring av hver aminosyre (symboler er definert i tabell 1).
MERK: Det kan ta flere minutter å generere justeringen. - For å se aminosyreegenskapene i farge, klikk på Vis farger-koblingen rett over sekvensene for å farge aminosyrene i henhold til deres egenskaper (definert i tabell 2).
- Kopier og lim inn sekvensjusteringen i et tekstbehandlings- eller lysbildefremvisningsprogram for å generere en figur- eller illustrasjonsfil (f.eks.
MERK: Bruk en monospaced skrift for justeringen, for eksempel Courier. - For å se andre utganger fra Clustal Omega-resultatsiden , klikk på de aktuelle kategoriene (dvs. guidetre eller fylogenetisk tre).
- Klikk kategorien Resultater Seere for alternativer for å vise sekvensinformasjonen ved hjelp av Jalview, et gratis program som spesialiserer seg på redigering, visualisering og analyse55 med flere sekvensjusteringer, eller for å få tilgang til direkte koblinger til MView og Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Her vil vi bruke den validerte mikroprotein mitoregulin (Mtln) som et eksempel for å demonstrere hvordan en konservert sORF vil generere en positiv PhyloCSF-poengsum som lett kan visualiseres og analyseres på UCSC Genome Browser. Mitoregulin ble tidligere kommentert som en ikke-koding RNA (tidligere humant gen ID LINC00116 og mus gen ID 1500011K16Rik). Komparative genomikk- og sekvensbevaringsanalysemetoder spilte en kritisk rolle i den første oppdagelsen 40,57,58,59,60,61, og fremhevet styrken til disse metodene. I dette eksemplet brukes musen GRCm38/mm10 (des. 2011). Søket kan utføres ved hjelp av genidentifikatorene (mitoregulin, Mtln) eller genposisjonen (chr2:127,791,364-127,792,496) som beskrevet i protokoll § 2. Alternativt kan aminosyresekvensen for mitoregulin (vist i figur 2) søkes ved hjelp av BLAT-verktøyet (beskrevet i protokoll avsnitt 3).
En skjerm som ligner på den som er avbildet i figur 1A , vises med PhyloCSF Track Hub synlig øverst på skjermen. Smoothed PhyloCSF-sporene (utjevnet med en skjult Markov-modell som definerer en sannsynlighet for at hver codon koder) er avbildet som seks spor totalt, med tre spor som tilsvarer plussstrengen av DNA (avbildet i grønt som PhyloCSF +1, +2 og +3) og tre spor som tilsvarer minusstrengen av DNA (avbildet i rødt som PhyloCSF -1, -2 og -3). Disse sporene representerer de tre potensielle leserammene for genet av interesse i hver retning. I nettleservinduet er exons avbildet som blå rektangler forbundet med tynne blå horisontale linjer, som representerer intronene. Pilspissene på de introniske regionene indikerer hvilken retning genet transkribereres i (og dermed hvilken tråd å fokusere på for PhyloCSF-poengsummen). For eksempelet med Mtln i figur 1 peker de introniske pilspissene mot venstre. Derfor transkriberes Mtln-genet fra minusstrengen av DNA, og den relevante PhyloCSF-poengsummen er avbildet i -1, -2 og -3 spor (i rødt).
Hvert PhyloCSF-spor er avbildet som en tynn svart linje med negative poengregioner avbildet i lysegrønn/ rødt under linjen og positive poengregioner angitt i mørkegrønn / rød over linjen. Som beskrevet i introduksjonen indikerer en positiv PhyloCSF-poengsum et bevart område som sannsynligvis koder. Legg merke til at for proteinkodingsregioner med spesielt høy sekvensbevaring, scorer de ofte også positivt på antisense-strengen; Imidlertid er PhyloCSF-poengsummen vanligvis høyere på riktig tråd. For eksempel kan dette ses i figur 1 for Mtln der riktig kodesekvens scorer veldig høyt i PhyloCSF -1-sporet, og antisense-strengen (PhyloCSF +2-sporet) genererer også en positiv poengsum. Som vist i figur 1A (indikert med svart boks), er det en region i den første eksonen av Mtln som scorer veldig høyt på PhyloCSF -1-sporet, noe som tyder på at dette kan tilsvare en kodingsregion. Hvis du vil undersøke dette området mer detaljert, er det nyttig å zoome inn og forstørre området (figur 1B). Som vist i figur 1C,D, begynner den positivt scorende regionen i den første eksonen av Mtln direkte over en startkodon (figur 1C) og avsluttes ved en stoppkodon (figur 1D), noe som indikerer at denne ORF er svært bevart og sterkt antyder at det er en koding ORF. Siden Mtln er på minus dna-strengen, vises start- og stoppkokonene som det omvendte komplementet til codonen (dvs. ATG-startkodonen vises som CAT [Figur 1C] og TGA-stoppkodon vises som TCA [Figur 1D]).
I tillegg til å bruke PhyloCSF til å søke etter konserverte regioner med mikroproteinkodingspotensial, kan denne teknikken også brukes som en første-pass analyse av putative noncoding RNAer for å utelukke tilstedeværelsen av en bevart ORF, og dermed gi støtte for en ikke-kodingsmerknad. For eksempel viser analyse av den godt karakteriserte lncRNA HOTAIR62,63 ved hjelp av PhyloCSF en negativ poengsum gjennom hele genet på tvers av alle seks sporene (figur 3), noe som sterkt indikerer mangel på sekvensbevaring og gir støtte for at HOTAIR er riktig kommentert som en ikke-koding RNA.
Som tydelig sett i figur 1, er hele kodingen ORF for mitoregulin plassert i en enkelt exon, og produserer dermed en enkel og grei avlesning av PhyloCSF med en enkelt, uavbrutt, positivt scoreregion. Imidlertid er PhyloCSF-sporhubdata ikke alltid så klare og enkle å tolke. For eksempel kan mitolamban/Stmp1/Mm47 mikroprotein kodet av musen 1810058I24Rik gen 47,64,65 viser en konservert ORF som spenner over tre exons (Figur 4A), og den positive PhyloCSF-poengsummen hopper fra +2-sporet i exon 1 (figur 4B) til +3-sporet i exon 2 (figur 4C), og deretter tilbake til +2-sporet i exon 3 (figur 4D ). Mens dette ved første øyekast ser forvirrende ut, er forklaringen ganske grei. PhyloCSF scorer de seks potensielle leserammene (tre på plussstrengen av DNA og tre på minusstrengen) av genomiske regioner uten å vurdere den spesifikke exon / intronarkitekturen for hvert gen. Derfor beholder den intronisk sekvensinformasjon i 3-nukleotid periodiciteten til leserammene. Således, hvis en intron inneholder en rekke nukleotider som ikke kan deles på tre (dvs. tre nukleotider / codon), vil PhyloCSF-leserammen hoppe fra ett spor til et annet.
Til slutt kan PhyloCSF også effektivt brukes til å identifisere flere distinkte kodings-ORFer i et enkelt RNA-molekyl. MIEF1-mikroproteinet (MIEF1-MP) er for eksempel kodet innenfor 5' UTR for mitokondrieforlengelsesfaktor 1 (MIEF1)66 (figur 5). Når MIEF1 genomisk region analyseres av PhyloCSF, kan en diskret positiv PhyloCSF-skår som tilsvarer MIEF1-MP (figur 5C) lett observeres oppstrøms for hoved-CD-ene for MIEF1 (figur 5B). Videre diskusjon om MIEF1 og tilhørende mikroprotein (MIEF1-MP) er gitt nedenfor i diskusjonen sammen med et sammendrag av styrker og svakheter ved metodene og protokollene som er skissert i denne artikkelen.

Figur 1: PhyloCSF-analyse av mitoregulin (Mtln)-genet indikerer en region med høy sekvensbevaring som tilsvarer et validert mikroprotein. (A) Skjermbilder av UCSC Genome Browser og PhyloCSF Tracks viser at Mtln inneholder to exons og en enkelt intron. Pilspissene i intronpunktet til venstre, som indikerer at Mtln-genet er transkribert fra minusstrengen av DNA, og de relevante PhyloCSF-poengsummene vises derfor i -1, -2 og -3 sporene (i rødt). Den komplette mitoregulinkodingssekvensen finnes i Exon 1 og scorer høyt på PhyloCSF -1-sporet (B). En konservert startkodon kan tydelig observeres i begynnelsen av den positivt scorende regionen i PhyloCSF -1-sporet (C), som er uthevet med en grønn boks (CAT, omvendt komplement ATG). I tillegg er en konservert stoppkodon (TCA, omvendt komplement TGA) indikert med en rød boks i panel (D), som samsvarer med slutten av den positivt scorende PhyloCSF-regionen. Detaljert informasjon om Mtln-genet finner du ved å klikke på Mtln-genidentifikatoren i den blå boksen (vist i panel A). Vær oppmerksom på at høyt bevarte proteinkodingsregioner ofte også scorer positivt på antisensestrengen (sett her i PhyloCSF +2-sporet for Mtln). PhyloCSF-poengsummen er imidlertid vanligvis høyere på riktig tråd (PhyloCSF -1-sporet i dette eksemplet). Klikk her for å se en større versjon av denne figuren.

Figur 2: Flere arter sekvens justering av mikroprotein mitoregulin generert ved hjelp av Clustal Omega program. Mitoregulin aminosyresekvensene for de åtte angitte artene ble ekstrahert som beskrevet i protokoll avsnitt 6 og justert med Clustal Omega multisekvensjusteringsverktøy. Egenskapene til aminosyrene er indikert etter farge (rød, liten/hydrofob, blå, sur; magenta, grunnleggende; grønn, hydroksl/sulhydryl/amin) (nærmere definert i tabell 2). Symbolene under aminosyrene indikerer graden av bevaring (stjerner, fullkonserverte rester; koloner, aminosyrer med sterkt like egenskaper; perioder, bevaring mellom grupper av svakt like egenskaper) (detaljert omfattende i tabell 1). Klikk her for å se en større versjon av denne figuren.
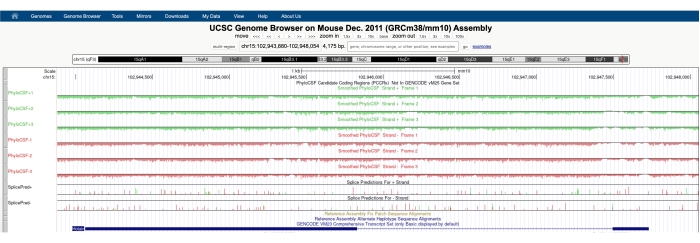
Figur 3: Et skjermbilde av PhyloCSF-sporene for den validerte lange, ikke-kodede RNA Hotair viser mangel på sekvensbevaring gjennom hele det genomiske locus. Pilspissene i den introniske regionen Hotair peker mot venstre, noe som indikerer at lncRNA er transkribert fra den negative dna-strengen, og derfor bør PhyloCSF -1, -2 og -3 spor være fokus for analyse. Legg merke til at PhyloCSF-poengsummen er negativ gjennom hele genet (for alle seks sporene), noe som indikerer mangel på sekvensbevaring, som støtter riktig merknad som en ikke-koding av RNA. Klikk her for å se en større versjon av denne figuren.

Figur 4: PhyloCSF-analyse av mus 1810058I24Rik-genet , som koder mikroprotein mitolamban/Stmp1/Mm47. (A) Musen 1810058I24Rik-genet består av tre eksoner, og pilspissene i de introniske områdene peker til høyre, noe som indikerer at det transkriberes på plussstrengen av DNA og derfor bør PhyloCSF +1, +2 og +3 spor analyseres. Den bevarte mikroproteinkodingssekvensen spenner over alle tre exons, som starter i exon 1 (B), leser gjennom exon 2 (C), og slutter på exon 3 (D). Legg merke til at den positive PhyloCSF-poengsummen finnes på +2-sporet i exon 1, +3-sporet i exon 2 og +2-sporet i exon 1. Årsaken til bevegelsen av den positive poengsummen fra det ene sporet til det andre er at PhyloCSF analyserer de seks potensielle leserammene i DNA-sekvensen uavhengig av genets ekson/ intronstruktur. Derfor vil en intron som inneholder en rekke nukleotider som ikke kan deles på tre (tre nukleotider/codon) føre til et skifte i leserammen til et annet spor. Klikk her for å se en større versjon av denne figuren.

Figur 5: Analyse av mief1 genomisk locus med PhyloCSF identifiserer en region med proteinkodingspotensial i 5' UTR som er uavhengig av de viktigste Mief1 CDS på den delte RNA. Dette konserverte oppstrøms ORF (uORF) har vist seg å kode et mikroprotein kalt Mief1-MP. (A) Oversikt over Mief1 genomisk locus. Pilspissene i intronene peker mot høyre, noe som indikerer at Mief1 er transkribert fra plussstrengen av DNA (fokus på PhyloCSF +1, +2 og +3 spor for å bestemme kodingspotensialet). Den viktigste Mief1 CDS koder en 463 aminosyre protein og er vist i panel (B). Imidlertid er det også et tydelig konservert oppstrøms ORF innenfor 5' UTR av Mief1 som koder et unikt 70 aminosyremikroprotein kalt Mief1-MP (C). Som sett i panel C, har Mief1-MP sin egen bevarte start og stopp codon i Mief1 5 ' UTR, og ORF scorer veldig høyt på PhyloCSF +1-sporet, og gir sterke bevis på at det koder et funksjonelt mikroprotein. Forkortelser: ORF = åpen leseramme; uORF = oppstrøms ORF; UTR = uoversatt område; CDS = kodesekvens. Klikk her for å se en større versjon av denne figuren.
| Symbol | Nivå av Amino Acid Conservation | Grupperte aminosyrer |
| Stjerne (*) | Fullt bevarte rester | Gjelder ikke (enkeltreserverte rester) |
| Kolon (:) | Grupper med svært like egenskaper | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FAKTISK VEKT |
| Periode (.) | Grupper med svakt like egenskaper | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Mellomrom (ikke noe symbol) | Ingen likhet | Gjelder ikke (ingen likhet) |
Tabell 1: Definisjoner av konsensussymboler for multisekvensjusteringer generert av Clustal Omega. Sekvensjusteringen av flere arter vist i figur 2 ble generert ved hjelp av Clustal Omega52. Forkortelser: serin (S), treonin (T), alanin (A), asparagin (N), glutaminsyre (E), glutamin (Q), lysin (K), asparaginsyre (D), arginin (R), metionin (M), er solanin (I), leucin (L), fenylalanin (F), histidin (H), tyrosin (Y), tryptofan (W), cystein (C), valin (V), glycin (G), prolin (P).
| Skriftfarge | Eiendom | Aminosyrerester [forkortelse] |
| Rød | Liten, hydrofob | alanin [A], valin [V], fenylalanin [F], prolin [P], metionin [M], isoleucin [I], leucin [L], tryptofan [W] |
| Blå | Sur | asparaginsyre [D], glutaminsyre [E] |
| Magenta | Grunnleggende | arginin [R], lysin [K] |
| Grønn | Hydroksl, sulhydryl, amin, +G | serin [S], treonin [T], tyrosin [Y], histidin [H], cystein [C], asparagin [N], glycin [G], glutamin [Q] |
Tabell 2: Egenskaper av aminosyrene avbildet i figur 2. Clustal Omega52 ble brukt til å generere multisekvensjusteringen vist i figur 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Protokollen som presenteres her gir detaljerte instruksjoner om hvordan du forhører genomiske interesseregioner for mikroproteinkodingspotensial ved hjelp av PhyloCSF på den brukervennlige UCSC Genome Browser 48,49,50,51. Som beskrevet ovenfor er PhyloCSF en kraftig komparativ genomisk algoritme som integrerer fylogenetiske modeller og kodonerstatningsfrekvenser for å identifisere evolusjonære signaturer som er typiske for proteinkodingsgener 48,49. PhyloCSF har blitt mye brukt til å identifisere funksjonelle mikroproteiner i genomiske regioner som tidligere ble kommentert som ikke-koding 38,39,40,41,42,43,44,45,46,47 , og denne tilnærmingen har vist seg å overgå andre komparative genomiske metoder for korte sekvenser som mikroproteiner så små som 13 aminosyrer og for små eksoniske eksoniske proteiner 35,48,49. Spesielt strekker nytten av PhyloCSF som en robust metode for å identifisere funksjonelle proteinkodingssekvenser via evolusjonær bevaring utover vertebrat- og hvirvelløse arter og har til og med nylig blitt brukt på virale genomer for å lykkes med å forhøre proteinkodingskapasiteten til SARS-CoV-2-genomet67.
I tillegg til å identifisere putative kodesekvenser i kommenterte ikke-kodende RNAer, en fordel med PhyloCSF er at den også på en pålitelig måte kan oppdage konserverte mikroproteiner kodet av ORFer innenfor kommenterte utranslaterte regioner (UTRs) av kanoniske proteinkodingsgener, inkludert både 5' oppstrøms og 3' nedstrøms ORF-er (henholdsvis uORF-er og dORFer)8,19,66,68 . For eksempel er MIEF1 mikroprotein (MIEF1-MP) kodet i 5' UTR av mitokondrieforlengelsesfaktor 1 (MIEF1)66. Når det gjelder MIEF1-MP, observeres en diskret positiv PhyloCSF-poengsum som tilsvarer MIEF1-MP oppstrøms for ORF som koder MIEF1 (figur 5). Mens noen uORF-kodede mikroproteiner samhandler direkte med nedstrøms kanoniske proteiner på deres delte mRNA, (f.eks. MIEF1-MP og MIEF1), fungerer andre uavhengig av proteinet som er kodet av hoved-CDS66,68. Derfor, når du karakteriserer uORF-kodede mikroproteiner, bør det ikke antas at de fungerer via direkte regulering av deres nedstrøms proteinprodukt.
Mens PhyloCSF har mange klare styrker som et verktøy for identifisering av bevarte mikroproteinkodingssekvenser, er det viktig å gjenkjenne flere begrensninger i denne metoden. For det første, mens sekvensbevaring sterkt antyder at en genomisk region har gjennomgått funksjonelt utvalg og dermed koding, utelukker mangel på robust bevaring og en resulterende negativ PhyloCSF-poengsum ikke definitivt kodingspotensial for en gitt sekvens. Med andre ord, å stole utelukkende på PhyloCSF kan resultere i tilsyn med oversatte ORFer som ikke er sterkt bevart, men som fortsatt produserer funksjonelle mikroproteiner. Spesielt kan genomiske regioner med lav bevaring eller negative bevaringspoeng tilsvare artsspesifikke koderegioner eller de av evolusjonære "unge" gener via sekvensforskjell eller de novo genfødsel 46,69,70,71,72,73,74. For eksempel blir mikroproteinet ASAP, som er kodet av det som tidligere ble antatt å være den menneskelige noncoding RNA LINC00467, ikke scoret positivt av PhyloCSF fordi aminosyresekvensen bare er bevart hos høyere pattedyr75. I tillegg identifiserte nyere studier flere menneskespesifikke mikroproteiner, inkludert en kodet av den intergene lncRNA RP3-527G5.1, som ikke genererer en positiv PhyloCSF-poengsum68,72. I denne forbindelse kan fraværet av en positiv PhyloCSF-poengsum ikke tolkes som bevis på et ikke-kodingsområde og bør tolkes med forsiktighet.
En annen vurdering å huske på når du bruker PhyloCSF er at selv om en positiv poengsum er svært antydende for funksjonelt utvalg og proteinkodingskapasitet, kan denne bevislinjen ikke stå alene og må valideres eksperimentelt. Eksempler på metoder som kan brukes til å generere støttende bevis for stabilt mikroproteinuttrykk inkluderer påvisning av det putative proteinet ved massespektrometri eller vestlig blotting ved hjelp av et antistoff hevet mot mikroproteinsekvensen av interesse. Alternativt, siden det kan være utfordrende å generere pålitelige antistoffer for mikroproteiner på grunn av mangel på sekvensvalg for optimal antigenitet, er det også mulig å bruke CRISPR / Cas9 og den homologistyrte reparasjonsveien (HDR) for å introdusere en epitopmerke i den endogene locus i ramme med den putative mikroproteinsekvensen, og dermed lette påvisning av proteinet av interesse ved hjelp av et høyt affinitets antistoff (f.eks. FLAGG, HA, V5, Myc)18. En endelig begrensning av PhyloCSF å erkjenne er at selv om det for tiden er integrert i mange av de ofte brukte genomiske forsamlinger, inkludert Homo sapiens (human hg19, hg38), Mus musculus (mus mm10, mm39), Gallus gallus (kylling, galGal4, galGal6), Drosophila melanogaster (fruktflue, dm6), Caenorhabditis elegans (nematoder, ce11) og SARS-CoV-2 (wuhCor1), er det fortsatt mange arter som for tiden ikke kan spørres direkte på UCSC Genome Browser.
Identifisering av konserverte domener eller sekvenskarakteristikker i identifiserte mikroproteiner kan bidra til å øke tilliten til deres funksjonelle relevans og gi litt innsikt i deres putative funksjon. Her gir vi anbefalinger for spesifikke verktøy og ressurser som kan brukes til å analysere identifiserte mikroprotein aminosyresekvenser nærmere for å få slik innsikt. De spesifikke verktøyene som er oppført nedenfor (og oppsummert i materialtabellen) er fritt tilgjengelige for publikum, og vi har funnet at de er spesielt brukervennlige og robuste i mikroproteinstudier 18,38,39,40,41,47. Utover verktøyene som er beskrevet her, finnes det en rekke ekstra ressurser som finnes i bioinformatikkressursportaler som Expasy (https://www.expasy.org) og EMBL-EBI (https://www.ebi.ac.uk/services/all). Detaljering av detaljene for hvert av verktøyene i disse repositoriene er imidlertid utenfor omfanget av denne artikkelen. Her anbefaler vi følgende ressurser.
For det første analyserer TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) proteinsekvenser av interesse for tilstedeværelse av transmembrandomener. Spesielt inneholder en rekke mikroproteiner som har blitt funksjonelt karakterisert så langt, single-pass transmembrane domener, noe som letter lokaliseringen til membranregioner og muliggjør deres direkte regulering av ionkanaler, vekslere og membranrelaterte enzymer30. For det andre er National Center for Biotechnology Information (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) et populært verktøy som brukes til å identifisere bevarte domener innen protein- eller kodingskjernepunktsekvenser. For det tredje gir Protein-familien (Pfam)78-databasen (http://pfam.xfam.org) justeringer og klassifiseringer av proteinfamilier og domener. For det fjerde er WoLF PSORT79 (https://wolfpsort.hgc.jp/) et verktøy som kan brukes til å forutsi subcellulær proteinlokalisering. For det femte er COXPRESdB80 en gen-co-expression database (https://coxpresdb.jp) som gir koregulerte genrelasjoner for å estimere genfunksjoner. Til slutt er SignalP 6.081 et mye brukt prediksjonsprogram (https://services.healthtech.dtu.dk/service.php?SignalP) som gjenkjenner tilstedeværelsen av en signalpeptidsekvens og forutsier plasseringen av spaltingsstedet.
Oppsummert kan metodene beskrevet her brukes til effektivt å analysere genomiske interesseområder for proteinkodingspotensial ved hjelp av PhyloCSF på UCSC Genome Browser. Disse metodene er svært tilgjengelige og kan enkelt læres og brukes effektivt av enkeltpersoner uten forutgående opplæring eller kompetanse innen bioinformatikk eller komparativ genomikk. Som vist her i detalj, er PhyloCSF et kraftig verktøy som kan brukes som en første-pass analyse for å bidra til å skille proteinkoding versus ikke-koding gener i virveldyr, hvirvelløse og virale genomer, og styrkene til denne tilnærmingen oppveier sterkt de nevnte svakhetene.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne erklærer at de ikke har konkurrerende økonomiske interesser.
Acknowledgments
Dette arbeidet ble støttet av tilskudd fra National Institutes of Health (HL-141630 og HL-160569) og Cincinnati Children's Research Foundation (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
