

Summary
Protokollet som beskrivs här ger detaljerade instruktioner om hur man analyserar genomiska regioner av intresse för mikroproteinkodningspotential med hjälp av PhyloCSF i den användarvänliga UCSC Genome Browser. Dessutom rekommenderas flera verktyg och resurser för att ytterligare undersöka sekvensegenskaper hos identifierade mikroproteiner för att få insikt i deras förmodade funktioner.
Abstract
Nästa generations sekvensering (NGS) har drivit genomikområdet framåt och producerat helgenomsekvenser för många djurarter och modellorganismer. Men trots denna mängd sekvensinformation har omfattande genannoteringsinsatser visat sig vara utmanande, särskilt för små proteiner. I synnerhet utformades konventionella proteinannoteringsmetoder för att avsiktligt utesluta förmodade proteiner kodade av korta öppna läsramar (sORF) mindre än 300 nukleotider i längd för att filtrera bort det exponentiellt högre antalet falska icke-kodande sORF i hela genomet. Som ett resultat har hundratals funktionella små proteiner som kallas mikroproteiner (<100 aminosyror i längd) felaktigt klassificerats som icke-kodande RNA eller förbisetts helt.
Här tillhandahåller vi ett detaljerat protokoll för att utnyttja gratis, offentligt tillgängliga bioinformatiska verktyg för att fråga genomiska regioner efter mikroproteinkodningspotential baserat på evolutionär bevarande. Specifikt tillhandahåller vi steg-för-steg-instruktioner om hur man undersöker sekvensbevarande och kodningspotential med hjälp av fylogenetiska kodonersättningsfrekvenser (PhyloCSF) på den användarvänliga University of California Santa Cruz (UCSC) Genome Browser. Dessutom beskriver vi steg för att effektivt generera flera arters inriktningar av identifierade mikroproteinsekvenser för att visualisera aminosyrasekvensbevarande och rekommenderar resurser för att analysera mikroproteinegenskaper, inklusive förutsagda domänstrukturer. Dessa kraftfulla verktyg kan användas för att identifiera förmodade mikroproteinkodande sekvenser i icke-kanoniska genomiska regioner eller för att utesluta närvaron av en konserverad kodningssekvens med translationell potential i ett icke-kodande transkript av intresse.
Introduction
Identifieringen av den fullständiga uppsättningen kodande element i genomet har varit ett viktigt mål sedan initieringen av Human Genome Project och är fortfarande ett centralt mål mot förståelsen av biologiska system och etiologin för genetiska baserade sjukdomar 1,2,3,4. Framsteg inom NGS-tekniker har lett till produktion av helgenomsekvenser för ett stort antal organismer, inklusive ryggradsdjur, ryggradslösa djur, jäst och växter5. Dessutom har transkriptionella sekvenseringsmetoder med hög genomströmning ytterligare avslöjat komplexiteten hos det cellulära transkriptomet och identifierat tusentals nya RNA-molekyler med både proteinkodande och icke-kodande funktioner 6,7. Avkodning av denna stora mängd sekvensinformation är en pågående process, och utmaningar kvarstår med omfattande genannoteringsinsatser8.
Den senaste utvecklingen av translationella profileringsmetoder, inklusive ribosomprofilering 9,10 och poly-ribosomsekvensering11, har gett bevis som tyder på att hundratals icke-kanoniska översättningshändelser kartläggs till för närvarande oannoterade SORF i hela genomet, med potential att generera små proteiner som kallas mikroproteiner eller mikropeptider 12,13,14,15,16, 17. Mikroproteiner har framstått som en ny klass av mångsidiga proteiner som tidigare förbisetts av vanliga genannoteringsmetoder på grund av deras lilla storlek (<100 aminosyror) och brist på klassiska proteinkodande genegenskaper 8,12,18,19,20. Mikroproteiner har beskrivits i praktiskt taget alla organismer, inklusive jäst 21,22, flugor 17,23,24 och däggdjur 25,26,27,28, och har visat sig spela kritiska roller i olika processer, inklusive utveckling, metabolism och stresssignalering 19,20,29, 30,31,32,33,34. Således är det absolut nödvändigt att fortsätta att bryta genomet för ytterligare medlemmar av denna länge förbisedda klass av funktionella små proteiner.
Trots det utbredda erkännandet av mikroproteinernas biologiska betydelse är denna klass av gener fortfarande kraftigt underrepresenterad i genomanteckningar, och deras exakta identifiering fortsätter att vara en pågående utmaning som har hindrat framsteg inom området. Olika beräkningsverktyg och experimentella metoder har nyligen utvecklats för att övervinna svårigheterna med att identifiera mikroproteinkodningssekvenser (diskuteras utförligt i flera omfattande recensioner 8,35,36,37). Många nyligen genomförda mikroproteinidentifieringsstudier 38,39,40,41,42,43,44,45,46,47 har förlitat sig starkt på användningen av en sådan algoritm som heter PhyloCSF 48,49 , en kraftfull jämförande genomisk metod som kan utnyttjas för att skilja konserverade proteinkodande regioner i genomet från de som inte är kodande.
PhyloCSF jämför kodonsubstitutionsfrekvenser (CSF) med hjälp av nukleotidjusteringar med flera arter och fylogenetiska modeller för att detektera evolutionära signaturer av proteinkodande gener. Detta empiriska modellbaserade tillvägagångssätt bygger på förutsättningen att proteiner primärt bevaras på aminosyranivå snarare än nukleotidsekvensen. Därför poängsätts synonyma kodonsubstitutioner, som kodar för samma aminosyra, eller kodonsubstitutioner till aminosyror med konserverade egenskaper (dvs laddning, hydrofobicitet, polaritet) positivt, medan icke-synonyma substitutioner, inklusive missense och nonsenssubstitutioner, får negativt poäng. PhyloCSF är utbildad på helgenomdata och har visat sig vara effektiv för att poängsätta korta delar av en kodningssekvens (CDS) isolerat från hela sekvensen, vilket är nödvändigt vid analys av mikroproteiner eller enskilda exoner av standardproteinkodande gener48,49.
I synnerhet gör den senaste integrationen av PhyloCSF-spårnaven i University of California Santa Cruz (UCSC) Genome Browser 49,50,51 det möjligt för utredare av alla bakgrunder att enkelt komma åt ett användarvänligt gränssnitt för att fråga genomiska regioner av intresse för proteinkodningspotential. Protokollet som beskrivs nedan ger detaljerad instruktion om hur man laddar PhyloCSF-spårnaven på UCSC Genome Browser och därefter förhör genomiska regioner av intresse för att söka efter proteinkodande regioner med hög konfidens (eller bristen på det). Dessutom, i det fall där en positiv PhyloCSF-poäng observeras, avgränsas steg för att ytterligare analysera mikroproteinkodningspotential och effektivt generera flera arters inriktningar av de identifierade aminosyrasekvenserna för att illustrera bevarande av sekvenser mellan arter. Slutligen introduceras flera ytterligare offentligt tillgängliga resurser och verktyg i diskussionen för att kartlägga identifierade mikroproteinegenskaper, inklusive förutsagda domänstrukturer och insikt i förmodad mikroproteinfunktion.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Protokollet som beskrivs nedan beskriver steg för att ladda och navigera i PhyloCSF-webbläsarspåren i UCSC Genome Browser (genererad av Mudge et al.49). För allmänna frågor om UCSC Genome Browser finns en omfattande användarhandbok för genomwebbläsare här: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Ladda PhyloCSF Track Hub till UCSC Genome Browser
- Öppna ett webbläsarfönster och navigera till UCSC Genome Browser (https://genome.ucsc.edu/).
- Under rubriken Våra verktyg väljer du alternativet Spåra hubbar .
Alternativet Track Hubs finns också under fliken Mina data . - På fliken Offentliga hubbar skriver du PhyloCSF i rutan Söktermer . Klicka på knappen Sök efter offentliga hubbar .
- Anslut till PhyloCSF genom att klicka på knappen Anslut för Hub Name PhyloCSF (Beskrivning: Evolutionär proteinkodningspotential mätt med PhyloCSF).
OBS: Denna Track Hub kommer att laddas till många enheter, inklusive människa (hg19 och hg38) och mus (mm10 och mm39). - När du har klickat på anslut väntar du på att omdirigeras till sidan UCSC Genome Browser Gateway (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Navigera till gener av intresse med hjälp av genidentifierare
- Välj den art och genomsammansättning som ska frågas. Om du vill fråga en annan art (t.ex. mus) väljer du den art som är av intresse under rubriken Bläddra/välj art genom att klicka på lämplig ikon, eller skriver in arten i textrutan som säger Ange art, vanligt namn eller sammansättnings-ID.
OBS: Sammansättningen listas direkt under rubriken Sök position . Vanligtvis är standardinställningen Mänsklig sammansättning (t.ex. december 2009 [GRCh37/hg19]). - Välj den sammansättning som ska sökas under rubriken Sök position med hjälp av rullgardinsmenyn.
- Ange position, gensymbol eller söktermer i rutan Position / sökterm och klicka på Gå för att navigera till en gen av intresse i genombläddraren.
- Om sökningen resulterade i flera matchningar väntar du på att omdirigeras till en sida som kräver val av en intresseposition. Klicka på lämplig gen av intresse.
3. Navigera till genomiska regioner av intresse med hjälp av sekvensinformation
- Navigera till UCSC Genome Browser (https://genome.ucsc.edu/) och välj BLAST-Like Alignment Tool (BLAT) under rubriken Våra verktyg för att fråga en specifik DNA- eller proteinsekvens. Alternativt kan du hålla markören över fliken Verktyg och välja Blat-alternativet eller följa den här länken: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Välj art (Genom) och Sammansättning av intresse med hjälp av rullgardinsmenyerna.
- Definiera frågetypen med hjälp av rullgardinsmenyn.
- Klistra in sekvensen av intresse i textrutan BLAT Search Genome och klicka på Skicka.
- Klicka på webbläsarlänken under rubriken ÅTGÄRDER för att navigera till det genomiska området av intresse.
4. Identifiera bevarade sORF med hjälp av PhyloCSF Track Data
- Skanna visuellt det genomiska området av intresse för att positivt poängsätta PhyloCSF-regioner (figur 1).
OBS: För en detaljerad förklaring av hur man visuellt tolkar PhyloCSF-poäng i UCSC Genome Browser, se avsnittet om representativa resultat nedan. - Använd zoomfunktionen för att förstora regioner av intresse för att undersöka sekvensegenskaper och söka efter start/stopp-kodon. Om du vill zooma in manuellt håller du ned skifttangenten och klickar och håller ned musknappen medan du drar längs intresseområdet. Alternativt kan du använda knapparna zooma in och zooma ut högst upp på sidan för att navigera (alternativen 1,5x, 3x, 10x eller baszoom är tillgängliga).
OBS: Innan du använder knapparna för att zooma in / zooma ut är det nödvändigt att flytta genen så att intresseområdet ligger mitt på skärmen. För att utföra denna åtgärd, klicka på bilden och dra den åt vänster eller höger för att flytta det genomiska området horisontellt efter önskemål eller använd flyttpilarna högst upp på sidan. - Zooma in tills nukleotidsekvensen (bassekvensen) är synlig.
OBS: Nukleotidsekvensen visas direkt ovanför +1 Utjämnad PhyloCSF-poäng. - Skanna visuellt nukleotidsekvensen nära början och slutet av de positivt poängsatta PhyloCSF-regionerna för att identifiera förmodade start (ATG) och stopp (TGA / TAA / TAG) kodoner.
OBS: Om genen av intresse är på minussträngen av DNA, kommer start- och stoppkodonerna att vara det omvända komplementet (dvs. CAT för startkodonet och TCA / TTA / CTA för stoppkodonet).
5. Visa homologa regioner i andra genom
- Håll musen över rubriken Visa högst upp på sidan och klicka på alternativet I andra genom (konvertera ).
- Definiera genomet av intresse med hjälp av rullgardinsmenyn under rubriken Nytt genom .
- Välj den genomiska sammansättningen av intresse med hjälp av rullgardinsmenyn under rubriken Ny sammansättning och klicka sedan på knappen Skicka .
- När webbläsaren returnerar en lista över regioner i den nya enheten med likhet klickar du på länken för kromosomposition för att navigera till det homologa området av intresse.
OBS: Procentandelen av totala baser (nukleotider) och spännvidden som täcks av regionen kommer att definieras för varje listad region. Ju högre andel matchande baser, desto högre är bevarandet för regionen av intresse. - Följ samma navigeringsstrategier som beskrivs i avsnitt 4 för att analysera sekvensen.
6. Generera sekvensjusteringar med flera arter för mikroproteiner av intresse
- Klicka på genen av intresse i GENCODE-spåret i UCSC Genome Browser (indikeras i figur 1A med en blå ruta) för att navigera till genbeskrivningssidan.
- Under rubriken Sekvens och länkar till verktyg och databaser klickar du på länken i tabellen som läser Andra arter FASTA.
- Klicka på rutorna som är associerade med de arter som är av intresse för att välja dem. Klicka på Skicka. Kopiera och klistra in sekvenserna som visas längst ner på sidan i FASTA-format i ett ordbehandlingsdokument.
- Öppna ett andra webbläsarfönster och navigera till Clustal Omega Multiple Sequence Alignment tool52 på European Bioinformatics Institute (EMBL-EBI) webbplats53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Klistra in sekvensfilerna som fortfarande finns på Urklipp i rutan i STEG 1 som läser sekvenser i alla format som stöds. Bläddra till botten av sidan och klicka på Skicka. Titta under de justerade resultaten (i svart teckensnitt) för symboler som anger graden av bevarande av varje aminosyra (symboler definieras i tabell 1).
Det kan ta flera minuter att generera justeringen. - För att se aminosyraegenskaperna i färg, klicka på länken Visa färger direkt ovanför sekvenserna för att färga aminosyrorna enligt deras egenskaper (definieras i tabell 2).
- Kopiera och klistra in sekvensjusteringen i ett ordbehandlings- eller bildspelsprogram för att generera en figur- eller illustrationsfil (t.ex. figur 2).
Använd ett teckensnitt med monospace för justeringen, till exempel Courier. - För att se andra utdata från Clustal Omega-resultatsidan , klicka på lämpliga flikar (dvs . guideträd eller fylogenetiskt träd).
- Klicka på fliken Resultatvisare för alternativ för att visa sekvensinformationen med Jalview, ett gratis program som specialiserat sig på redigering, visualisering och analys av flera sekvensjusteringar55, eller för att få tillgång till direktlänkar till MView och Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Här kommer vi att använda det validerade mikroproteinet mitoregulin (Mtln) som ett exempel för att visa hur en bevarad sORF kommer att generera en positiv PhyloCSF-poäng som enkelt kan visualiseras och analyseras på UCSC Genome Browser. Mitoregulin kommenterades tidigare som ett icke-kodande RNA (tidigare humant gen-ID LINC00116 och musgen-ID 1500011K16Rik). Jämförande genomik och sekvensbevarande analysmetoder spelade en avgörande roll i dess första upptäckt 40,57,58,59,60,61, vilket belyser styrkan i dessa metoder. I det här exemplet används musens GRCm38/mm10-enhet (dec. 2011). Sökningen kan utföras med hjälp av genidentifierare (mitoregulin, Mtln) eller genpositionen (chr2:127,791,364-127,792,496) som beskrivs i protokollavsnitt 2. Alternativt kan aminosyrasekvensen för mitoregulin (visas i figur 2) sökas med blat-verktyget (beskrivs i protokollavsnitt 3).
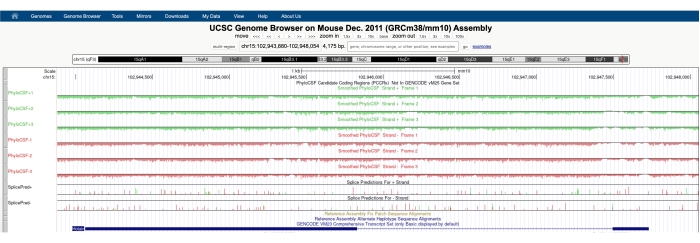
En skärm som liknar den som visas i figur 1A visas med PhyloCSF Track Hub synlig högst upp på skärmen. Smoothed PhyloCSF-spåren (utjämnade med en dold Markov-modell som definierar en sannolikhet för att varje kodon kodar) avbildas som sex totala spår, med tre spår som motsvarar DNA-strängen (avbildad i grönt som PhyloCSF +1, +2 och +3) och tre spår som motsvarar minussträngen av DNA (avbildad i rött som PhyloCSF -1, -2 och -3). Dessa spår representerar de tre potentiella läsramarna för genen av intresse i varje riktning. I webbläsarfönstret avbildas exoner som blå rektanglar förbundna med tunna blå horisontella linjer, som representerar intronerna. Pilspetsarna på de introniska regionerna indikerar vilken riktning genen transkriberas i (och därmed vilken sträng man ska fokusera på för PhyloCSF-poängen). För exemplet med Mtln i figur 1 pekar de introniska pilspetsarna åt vänster. Därför transkriberas Mtln-genen från dna-minussträngen, och den relevanta PhyloCSF-poängen avbildas i spåren -1, -2 och -3 (i rött).
Varje PhyloCSF-spår avbildas som en tunn svart linje med negativa poängregioner avbildade i ljusgrönt/rött under linjen och positiva poängområden som anges i mörkgrönt/rött ovanför linjen. Som beskrivs i inledningen indikerar en positiv PhyloCSF-poäng en bevarad region som sannolikt kodar. Observera att för proteinkodande regioner med särskilt hög sekvenskonservering får de ofta också positiva poäng på antisense-strängen; PhyloCSF-poängen är dock vanligtvis högre på rätt sträng. Detta kan till exempel ses i figur 1 för Mtln där rätt kodningssekvens får mycket höga poäng i PhyloCSF -1-spåret, och antisense-strängen (PhyloCSF +2-spåret) genererar också en positiv poäng. Som framgår av figur 1A (indikerad med svart låda) finns det en region i den första exonen av Mtln som får mycket höga poäng på PhyloCSF -1-spåret, vilket tyder på att detta kan motsvara en kodningsregion. För att undersöka denna region mer detaljerat är det bra att zooma in och förstora regionen (figur 1B). Som visas i figur 1C, D börjar det positivt poängsatta området i den första exonen av Mtln direkt över ett startkodon (figur 1C) och slutar vid ett stoppkodon (figur 1D), vilket indikerar att denna ORF är mycket bevarad och starkt tyder på att det är en kodande ORF. Eftersom Mtln är på minussträngen av DNA visas start- och stoppkodonerna som det omvända komplementet till kodonet (dvs. ATG-startkodonet visas som CAT [Figur 1C] och TGA-stoppkodonet visas som TCA [Figur 1D]).
Förutom att använda PhyloCSF för att söka efter konserverade regioner med mikroproteinkodningspotential, kan denna teknik också tillämpas som en första-pass-analys av förmodade icke-kodande RNA för att utesluta närvaron av en bevarad ORF, vilket ger stöd för en icke-kodande anteckning. Till exempel visar analys av det väl karakteriserade lncRNA HOTAIR 62,63 med PhyloCSF en negativ poäng genom hela genen över alla sex spåren (figur 3), vilket starkt indikerar brist på sekvensbevarande och ger stöd för att HOTAIR är korrekt kommenterad som ett icke-kodande RNA.
Som tydligt framgår av figur 1 ligger hela den kodande ORF för mitoregulin inom en enda exon, vilket ger en enkel och okomplicerad avläsning av PhyloCSF med en enda, oavbruten, positivt poänggivande region. PhyloCSF spårnavdata är dock inte alltid lika tydliga och lätta att tolka. Till exempel visar mitolamban / Stmp1 / Mm47 mikroprotein kodat av musen 1810058I24Rik-genen 47,64,65 en bevarad ORF som sträcker sig över tre exoner (figur 4A), och den positiva PhyloCSF-poängen hoppar från +2-spåret i exon 1 (figur 4B) till +3-spåret i exon 2 (figur 4C) och sedan tillbaka till +2-spåret i exon 3 (figur 4D ). Även om det vid första anblicken ser förvirrande ut, är förklaringen ganska enkel. PhyloCSF poängsätter de sex potentiella läsramarna (tre på plussträngen av DNA och tre på minussträngen) av genomiska regioner utan att ta hänsyn till den specifika exon / intronarkitekturen för varje gen. Därför behåller den den introniska sekvensinformationen i läsramarnas 3-nukleotid periodicitet. Således, om en intron innehåller ett antal nukleotider som inte är delbara med tre (dvs. tre nukleotider / kodon), kommer PhyloCSF-läsramen att hoppa från ett spår till ett annat.
Slutligen kan PhyloCSF också effektivt användas för att identifiera flera distinkta kodande ORF inom en enda RNA-molekyl. Till exempel kodas MIEF1-mikroproteinet (MIEF1-MP) inom 5 'UTR för mitokondriell töjningsfaktor 1 (MIEF1)66 (figur 5). När mief1-genomregionen analyseras av PhyloCSF kan en diskret positiv PhyloCSF-poäng motsvarande MIEF1-MP (figur 5C) lätt observeras uppströms huvud-CDS för MIEF1 (figur 5B). Ytterligare diskussion om MIEF1 och dess associerade mikroprotein (MIEF1-MP) ges nedan i diskussionen tillsammans med en sammanfattning av styrkorna och svagheterna i de metoder och protokoll som beskrivs i den här artikeln.

Figur 1: PhyloCSF-analys av mitoregulingenen (Mtln) indikerar en region med högsekvensbevarande motsvarande ett validerat mikroprotein. (A) Skärmdumpar av UCSC Genome Browser och PhyloCSF Tracks visar att Mtln innehåller två exoner och en enda intron. Pilspetsarna i intronen pekar åt vänster, vilket indikerar att Mtln-genen transkriberas från DNA-minussträngen, och de relevanta PhyloCSF-poängen visas därför i spåren -1, -2 och -3 (i rött). Den fullständiga mitoregulinkodningssekvensen finns i Exon 1 och får höga poäng på PhyloCSF -1-spåret (B). Ett bevarat startkodon kan tydligt observeras i början av det positivt poängsatta området i PhyloCSF -1-spåret (C), vilket markeras med en grön ruta (CAT, omvänd komplement ATG). Dessutom indikeras ett bevarat stoppkodon (TCA, omvänd komplement TGA) med en röd ruta i panelen (D), som överensstämmer med slutet av den positivt poängsatta PhyloCSF-regionen. Detaljerad information om Mtln-genen kan hittas genom att klicka på Mtln-genidentifieraren i den blå rutan (visas i panel A). Observera att mycket konserverade proteinkodande regioner ofta också får positiva poäng på antisense-strängen (ses här i PhyloCSF +2-spåret för Mtln). PhyloCSF-poängen är dock vanligtvis högre på rätt sträng (PhyloCSF -1-spåret i det här exemplet). Klicka här för att se en större version av denna siffra.

Figur 2: Flera arter sekvensjustering av mikroproteinet mitoregulin genererat med hjälp av Clustal Omega-programmet. Mitoregulinaminosyrasekvenserna för de åtta angivna arterna extraherades enligt beskrivningen i protokollavsnitt 6 och anpassades till Clustal Omega-verktyget för multipelsekvensjustering. Aminosyrornas egenskaper anges med färg (röd, liten/hydrofob; blå, sur; magenta, basisk; grön, hydroxl/sulfhydryl/amin) (närmare definierad i tabell 2). Symbolerna under aminosyrorna indikerar graden av bevarande (asterisker, helt konserverade rester; kolon, aminosyror med starkt liknande egenskaper; perioder, bevarande mellan grupper av svagt liknande egenskaper) (beskrivs utförligt i tabell 1). Klicka här för att se en större version av denna siffra.
Figur 3: En skärmdump av PhyloCSF-spåren för det validerade långa icke-kodande RNA Hotair visar brist på sekvensbevarande i hela dess genomiska locus. Pilspetsarna i den introniska regionen Hotair pekar åt vänster, vilket indikerar att lncRNA transkriberas från den negativa DNA-strängen, och därför bör PhyloCSF -1, -2 och -3 spåren vara i fokus för analysen. Observera att PhyloCSF-poängen är negativ genom hela genen (för alla sex spåren), vilket indikerar brist på sekvensbevarande, vilket stöder dess korrekta anteckning som ett icke-kodande RNA. Klicka här för att se en större version av denna siffra.

Figur 4: PhyloCSF-analys av musen 1810058I24Rik-genen , som kodar för mikroproteinet mitolamban / Stmp1 / Mm47. (A) Musen 1810058I24Rik-genen består av tre exoner, och pilspetsarna i de introniska regionerna pekar åt höger, vilket indikerar att den transkriberas på plussträngen av DNA och därför bör PhyloCSF +1-, +2- och +3-spåren analyseras. Den konserverade mikroproteinkodningssekvensen sträcker sig över alla tre exonerna, som börjar i exon 1 (B), läser igenom exon 2 (C) och slutar på exon 3 (D). Observera att den positiva PhyloCSF-poängen finns på +2-spåret i exon 1, +3-spåret i exon 2 och +2-spåret i exon 1. Anledningen till rörelsen av den positiva poängen från ett spår till ett annat är att PhyloCSF analyserar de sex potentiella läsramarna i DNA-sekvensen oberoende av genens exon / intronstruktur. Därför kommer en intron som innehåller ett antal nukleotider som inte är delbar med tre (tre nukleotider /kodon) att orsaka en förskjutning i läsramen till ett annat spår. Klicka här för att se en större version av denna siffra.

Figur 5: Analys av Mief1-genomisk locus med PhyloCSF identifierar en region med proteinkodande potential i 5 'UTR som är oberoende av den huvudsakliga Mief1 CDS på det delade RNA. Detta bevarade uppströms ORF (uORF) har visat sig koda för ett mikroprotein som heter Mief1-MP. (A) Översikt över Mief1 genomiska locus. Pilspetsarna i intronerna pekar åt höger, vilket indikerar att Mief1 transkriberas från DNA-strängen plus (fokusera på spåren PhyloCSF +1, +2 och +3 för att bestämma kodningspotentialen). Den huvudsakliga Mief1 CDS kodar för ett 463 aminosyraprotein och visas i panel (B). Det finns emellertid också ett distinkt bevarat uppströms ORF inom 5 'UTR för Mief1 som kodar för ett unikt 70 aminosyramikroprotein som kallas Mief1-MP (C). Som framgår av panel C har Mief1-MP sitt eget bevarade start- och stoppkodon inom Mief1 5 'UTR, och ORF får mycket höga poäng på PhyloCSF +1-spåret, vilket ger starka bevis för att det kodar för ett funktionellt mikroprotein. Förkortningar: ORF = öppen läsram; uORF = uppströms ORF; UTR = oöversatt region; CDS = kodningssekvens. Klicka här för att se en större version av denna siffra.
| Symbol | Nivå av aminosyra bevarande | Grupperade aminosyror |
| Asterisk (*) | Helt bevarade rester | Ej tillämpligt (enstaka, helt konserverade rester) |
| Kolon (:) | Grupper med starkt liknande egenskaper | Sta; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW |
| Period (.) | Grupper med svagt liknande egenskaper | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Blanksteg (ingen symbol) | Ingen likhet | Ej tillämpligt (ingen likhet) |
Tabell 1: Definitioner av konsensussymboler för flera sekvensjusteringar genererade av Clustal Omega. Den flera arter sekvensjustering som visas i figur 2 genererades med hjälp av Clustal Omega52. Förkortningar: serin (S), treonin (T), alanin (A), asparagin (N), glutaminsyra (E), glutamin (Q), lysin (K), asparaginsyra (D), arginin (R), metionin (M), isoleucin (I), leucin (L), fenylalanin (F), histidin (H), tyrosin (Y), tryptofan (W), cystein (C), valin (V), glycin (G), prolin (P).
| Teckenfärg | Egenskap | Aminosyrarest [Förkortning] |
| Röd | Liten, hydrofob | alanin [A], valin [V], fenylalanin [F], prolin [P], metionin [M], isoleucin [I], leucin [L], tryptofan [W] |
| Blå | Syrlig | asparaginsyra [D], glutaminsyra [E] |
| Magenta | Grundläggande | arginin [R], lysin [K] |
| Grön | Hydroxl, sulfhydryl, amin, +G | serin [S], treonin [T], tyrosin [Y], histidin [H], cystein [C], asparagin [N], glycin [G], glutamin [Q] |
Tabell 2: Egenskaper hos aminosyrorna som visas i figur 2. Clustal Omega52 användes för att generera multipelsekvensjusteringen som visas i figur 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Protokollet som presenteras här ger detaljerade instruktioner om hur man förhör genomiska regioner av intresse för mikroproteinkodningspotential med hjälp av PhyloCSF på den användarvänliga UCSC Genome Browser 48,49,50,51. Som beskrivits ovan är PhyloCSF en kraftfull jämförande genomikalgoritm som integrerar fylogenetiska modeller och kodonsubstitutionsfrekvenser för att identifiera evolutionära signaturer som är typiska för proteinkodande gener48,49. PhyloCSF har använts i stor utsträckning för att identifiera funktionella mikroproteiner i genomiska regioner som tidigare kommenterats som icke-kodande 38,39,40,41,42,43,44,45,46,47 , och detta tillvägagångssätt har visat sig överträffa andra jämförande genomikmetoder för korta sekvenser såsom mikroproteiner så små som 13 aminosyror och för små exoner av kanoniska proteiner 35,48,49. I synnerhet sträcker sig nyttan av PhyloCSF som en robust metod för att identifiera funktionella proteinkodande sekvenser via evolutionär bevarande utöver den för ryggradsdjur och ryggradslösa djurarter och har till och med nyligen tillämpats på virala genom för att framgångsrikt förhöra proteinkodningskapaciteten hos SARS-CoV-2-genomet67.
Förutom att identifiera förmodade kodningssekvenser inom kommenterade icke-kodande RNA, är en fördel med PhyloCSF att den också på ett tillförlitligt sätt kan detektera konserverade mikroproteiner kodade av ORF inom kommenterade oöversatta regioner (UTR) av kanoniska proteinkodande gener, inklusive både 5 'uppströms och 3' nedströms ORF (uORFs respektive dORF)8,19,66,68 . Till exempel kodas MIEF1-mikroproteinet (MIEF1-MP) i 5'UTR för mitokondriell töjningsfaktor 1 (MIEF1)66. När det gäller MIEF1-MP observeras en diskret positiv PhyloCSF-poäng som motsvarar MIEF1-MP uppströms ORF som kodar MIEF1 (figur 5). Medan vissa uORF-kodade mikroproteiner direkt interagerar med de nedströms kanoniska proteinerna på deras delade mRNA, (ex. MIEF1-MP och MIEF1), fungerar andra oberoende av proteinet som kodas av huvud-CDS66,68. Därför, när man karakteriserar uORF-kodade mikroproteiner, bör det inte antas att de fungerar via direkt reglering av deras nedströms proteinprodukt.
Medan PhyloCSF har många tydliga styrkor som ett verktyg för identifiering av konserverade mikroproteinkodande sekvenser, är det viktigt att känna igen flera begränsningar med denna metod. För det första, medan sekvensbevarande starkt tyder på att en genomisk region har genomgått funktionellt urval och därmed kodar, utesluter inte brist på robust bevarande och en resulterande negativ PhyloCSF-poäng definitivt kodningspotential för en given sekvens. Med andra ord kan uteslutande förlita sig på PhyloCSF resultera i övervakning av översatta ORF som inte är starkt bevarade men ändå producerar funktionella mikroproteiner. I synnerhet kan genomiska regioner med låg bevarande eller negativa bevarandepoäng motsvara artspecifika kodande regioner eller de av evolutionära "unga" gener via sekvensdivergens eller de novo-genfödelse 46,69,70,71,72,73,74. Till exempel är mikroproteinet ASAP, som kodas av vad som tidigare ansågs vara det mänskliga icke-kodande RNA LINC00467, inte positivt av PhyloCSF eftersom aminosyrasekvensen endast bevaras hos högre däggdjur75. Dessutom identifierade nyligen genomförda studier flera humanspecifika mikroproteiner, inklusive ett kodat av det intergeniska lncRNA RP3-527G5.1, som inte genererar en positiv PhyloCSF-poäng68,72. I detta avseende kan frånvaron av en positiv PhyloCSF-poäng inte tolkas som bevis på en icke-kodande region och bör tolkas med försiktighet.
En andra faktor att tänka på när du använder PhyloCSF är att även om en positiv poäng är mycket suggestiv för funktionellt urval och proteinkodningskapacitet, kan denna bevislinje inte stå ensam och måste valideras experimentellt. Exempel på metoder som kan användas för att generera stödjande bevis för stabilt mikroproteinuttryck inkluderar detektion av det förmodade proteinet genom masspektrometri eller western blotting med hjälp av en antikropp upphöjd mot mikroproteinsekvensen av intresse. Alternativt, eftersom det kan vara utmanande att generera tillförlitliga antikroppar för mikroproteiner på grund av bristen på sekvensval för optimal antigenicitet, är det också möjligt att använda CRISPR / Cas9 och den homologi-riktade reparationsvägen (HDR) för att införa en epitoptagg i den endogena locusen i ramen med den förmodade mikroproteinsekvensen, vilket underlättar detektionen av proteinet av intresse med hjälp av en antikropp med hög affinitet (t.ex. FLAGGA, HA, V5, Myc)18. En sista begränsning av PhyloCSF att erkänna är att även om det för närvarande är integrerat i många av de vanliga genomiska sammansättningarna, inklusive Homo sapiens (human hg19, hg38), Mus musculus (mus mm10, mm39), Gallus gallus (kyckling, galGal4, galGal6), Drosophila melanogaster (fruktfluga, dm6), Caenorhabditis elegans (nematoder, ce11) och SARS-CoV-2 (wuhCor1), det finns fortfarande många arter som för närvarande inte kan efterfrågas direkt i UCSC Genome Browser.
Identifieringen av bevarade domäner eller sekvensegenskaper inom identifierade mikroproteiner kan bidra till att öka förtroendet för deras funktionella relevans och ge viss inblick i deras förmodade funktion. Här ger vi rekommendationer för specifika verktyg och resurser som kan användas för att analysera identifierade mikroproteinaminosyrasekvenser mer detaljerat för att få sådan insikt. De specifika verktygen som listas nedan (och sammanfattas i materialförteckningen) är fritt tillgängliga för allmänheten, och vi har funnit att de är särskilt användarvänliga och robusta i mikroproteinstudier 18,38,39,40,41,47. Utöver de verktyg som beskrivs här finns det en mängd ytterligare resurser som finns i bioinformatikresursportaler som Expasy (https://www.expasy.org) och EMBL-EBI (https://www.ebi.ac.uk/services/all). Att beskriva detaljerna för vart och ett av verktygen i dessa lagringsplatsen ligger dock utanför omfånget för den här artikeln. Här rekommenderar vi följande resurser.
Först analyserar TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) proteinsekvenser av intresse för närvaron av transmembrandomäner. I synnerhet innehåller ett antal mikroproteiner som hittills har karakteriserats funktionellt enkelpasstransmembrandomäner, vilket underlättar deras lokalisering till membranregioner och möjliggör deras direkta reglering av jonkanaler, växlare och membranassocierade enzymer30. För det andra är National Center for Biotechnology Information (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) ett populärt verktyg som används för att identifiera konserverade domäner inom protein- eller kodande nukleotidsekvenser. För det tredje tillhandahåller proteinfamiljen (Pfam)78 databasen (http://pfam.xfam.org) anpassningar och klassificeringar av proteinfamiljer och domäner. För det fjärde är WoLF PSORT79 (https://wolfpsort.hgc.jp/) ett verktyg som kan användas för att förutsäga subcellulär proteinlokalisering. För det femte är COXPRESdB80 en gen-co-expression databas (https://coxpresdb.jp) som tillhandahåller samreglerade genrelationer för att uppskatta genfunktioner. Slutligen är SignalP 6.081 ett allmänt använt prediktionsprogram (https://services.healthtech.dtu.dk/service.php?SignalP) som känner igen närvaron av en signalpeptidsekvens och förutsäger platsen för klyvningsstället.
Sammanfattningsvis kan de metoder som beskrivs här användas för att effektivt analysera genomiska regioner av intresse för proteinkodningspotential med hjälp av PhyloCSF i UCSC Genome Browser. Dessa metoder är mycket tillgängliga och kan enkelt läras och effektivt tillämpas av individer utan tidigare utbildning eller expertis inom bioinformatik eller jämförande genomik. Som visas här i detalj är PhyloCSF ett kraftfullt verktyg som kan tillämpas som en första-pass-analys för att skilja proteinkodande kontra icke-kodande gener hos ryggradsdjur, ryggradslösa djur och virala genom, och styrkorna i detta tillvägagångssätt uppväger kraftigt de noterade svagheterna.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Författarna förklarar att de inte har några konkurrerande ekonomiska intressen.
Acknowledgments
Detta arbete stöddes av bidrag från National Institutes of Health (HL-141630 och HL-160569) och Cincinnati Children's Research Foundation (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
