

Summary
Das hier beschriebene Protokoll enthält detaillierte Anweisungen zur Analyse genomischer Regionen von Interesse auf Mikroprotein-kodierendes Potenzial mit PhyloCSF im benutzerfreundlichen UCSC Genome Browser. Darüber hinaus werden mehrere Werkzeuge und Ressourcen empfohlen, um die Sequenzeigenschaften identifizierter Mikroproteine weiter zu untersuchen und Einblicke in ihre mutmaßlichen Funktionen zu erhalten.
Abstract
Next-Generation-Sequencing (NGS) hat das Gebiet der Genomik vorangetrieben und ganze Genomsequenzen für zahlreiche Tierarten und Modellorganismen hervorgebracht. Trotz dieser Fülle an Sequenzinformationen haben sich umfassende Genannotationsbemühungen als schwierig erwiesen, insbesondere für kleine Proteine. Insbesondere wurden konventionelle Proteinannotationsmethoden entwickelt, um mutmaßliche Proteine absichtlich auszuschließen, die durch kurze offene Leserahmen (sORFs) mit einer Länge von weniger als 300 Nukleotiden kodiert werden, um die exponentiell höhere Anzahl falscher nichtkodierender sORFs im gesamten Genom herauszufiltern. Infolgedessen wurden Hunderte von funktionellen kleinen Proteinen, die Mikroproteine genannt werden (<100 Aminosäuren lang), fälschlicherweise als nicht-kodierende RNAs klassifiziert oder ganz übersehen.
Hier stellen wir ein detailliertes Protokoll zur Verfügung, um kostenlose, öffentlich verfügbare bioinformatische Werkzeuge zu nutzen, um genomische Regionen nach Mikroprotein-kodierendem Potenzial auf der Grundlage evolutionärer Konservierung abzufragen. Insbesondere bieten wir Schritt-für-Schritt-Anleitungen zur Untersuchung der Sequenzerhaltung und des Kodierungspotenzials mit phylogenetischen Codon-Substitutionsfrequenzen (PhyloCSF) im benutzerfreundlichen Genome Browser der University of California Santa Cruz (UCSC). Darüber hinaus beschreiben wir Schritte zur effizienten Generierung mehrerer Speziesausrichtungen identifizierter Mikroproteinsequenzen, um die Erhaltung der Aminosäuresequenz zu visualisieren, und empfehlen Ressourcen zur Analyse von Mikroproteineigenschaften, einschließlich vorhergesagter Domänenstrukturen. Diese leistungsstarken Werkzeuge können verwendet werden, um mutmaßliche Mikroprotein-kodierende Sequenzen in nicht-kanonischen genomischen Regionen zu identifizieren oder das Vorhandensein einer konservierten kodierenden Sequenz mit translationalem Potenzial in einem nicht-kodierenden Transkript von Interesse auszuschließen.
Introduction
Die Identifizierung des gesamten Satzes kodierender Elemente im Genom ist seit Beginn des Humangenomprojekts ein wichtiges Ziel und bleibt ein zentrales Ziel für das Verständnis biologischer Systeme und die Ätiologie genetisch bedingter Krankheiten 1,2,3,4. Fortschritte in der NGS-Technik haben zur Produktion ganzer Genomsequenzen für eine große Anzahl von Organismen geführt, darunter Wirbeltiere, Wirbellose, Hefen und Pflanzen5. Darüber hinaus haben transkriptionelle Hochdurchsatz-Sequenzierungsmethoden die Komplexität des zellulären Transkriptoms weiter aufgedeckt und Tausende neuartiger RNA-Moleküle mit proteinkodierenden und nicht-kodierenden Funktionen identifiziert 6,7. Die Entschlüsselung dieser riesigen Menge an Sequenzinformationen ist ein fortlaufender Prozess, und die Herausforderungen bei umfassenden Genannotationsbemühungenbleiben bestehen 8.
Die jüngste Entwicklung translationaler Profilierungsmethoden, einschließlich Ribosomenprofilierung 9,10 und Polyribosomensequenzierung 11, hat Hinweise darauf geliefert, dass Hunderte von nichtkanonischen Translationsereignissen derzeit unannotierten sORFs im gesamten Genom zugeordnet sind, mit dem Potenzial, kleine Proteine zu erzeugen, die Mikroproteine oder Mikropeptide genannt werden 12,13,14,15,16, 17. Mikroproteine haben sich aufgrund ihrer geringen Größe (<100 Aminosäuren) und des Fehlens klassischer proteinkodierender Geneigenschaften 8,12,18,19,20 bisher von Standard-Genannotationsmethoden übersehen. Mikroproteine wurden in praktisch allen Organismen beschrieben, einschließlich Hefe 21,22, Fliegen 17,23,24 und Säugetieren 25,26,27,28, und es wurde gezeigt, dass sie eine entscheidende Rolle in verschiedenen Prozessen spielen, einschließlich Entwicklung, Stoffwechsel und Stresssignalisierung 19,20,29, 30,31,32,33,34. Daher ist es unerlässlich, das Genom weiterhin nach zusätzlichen Mitgliedern dieser lange übersehenen Klasse funktioneller kleiner Proteine zu durchsuchen.
Trotz der weit verbreiteten Anerkennung der biologischen Bedeutung von Mikroproteinen ist diese Klasse von Genen in Genomannotationen nach wie vor stark unterrepräsentiert, und ihre genaue Identifizierung ist nach wie vor eine ständige Herausforderung, die den Fortschritt auf diesem Gebiet behindert hat. Verschiedene Berechnungswerkzeuge und experimentelle Methoden wurden kürzlich entwickelt, um die Schwierigkeiten zu überwinden, die mit der Identifizierung von Mikroprotein-kodierenden Sequenzen verbunden sind (ausführlich diskutiert in mehreren umfassenden Übersichtsarbeiten 8,35,36,37). Viele neuere Mikroprotein-Identifizierungsstudien 38,39,40,41,42,43,44,45,46,47 haben sich stark auf die Verwendung eines solchen Algorithmus namens PhyloCSF verlassen 48,49 , ein leistungsstarker vergleichender Genomikansatz, der genutzt werden kann, um konservierte proteinkodierende Regionen des Genoms von denen zu unterscheiden, die nicht kodierend sind.
PhyloCSF vergleicht Codon-Substitutionshäufigkeiten (CSF) unter Verwendung von Multispezies-Nukleotid-Alignments und phylogenetischen Modellen, um evolutionäre Signaturen von proteinkodierenden Genen nachzuweisen. Dieser empirische modellbasierte Ansatz beruht auf der Prämisse, dass Proteine in erster Linie auf der Aminosäureebene und nicht auf der Nukleotidsequenz konserviert werden. Daher werden synonyme Codon-Substitutionen, die die gleiche Aminosäure kodieren, oder Codon-Substitutionen zu Aminosäuren mit konservierten Eigenschaften (dh Ladung, Hydrophobie, Polarität) positiv bewertet, während nicht-synonyme Substitutionen, einschließlich Missverständnisse und Unsinnssubstitutionen, negativ abschneiden. PhyloCSF wird auf Daten des gesamten Genoms trainiert und hat sich als wirksam erwiesen, um kurze Teile einer kodierenden Sequenz (CDS) isoliert von der vollständigen Sequenz zu bewerten, was bei der Analyse von Mikroproteinen oder einzelnen Exons von Standardprotein-kodierenden Genenerforderlich ist 48,49.
Insbesondere die jüngste Integration der PhyloCSF-Track-Hubs in den Genome Browser 49,50,51 der University of California Santa Cruz (UCSC) ermöglicht es Ermittlern aller Hintergründe, leicht auf eine benutzerfreundliche Oberfläche zuzugreifen, um genomische Regionen von Interesse nach Proteinkodierungspotenzial abzufragen. Das unten beschriebene Protokoll enthält detaillierte Anweisungen zum Laden der PhyloCSF-Track-Hubs im UCSC Genome Browser und zur anschließenden Abfrage genomischer Regionen, die für die Suche nach proteinkodierenden Regionen mit hohem Konfidenzniveau (oder deren Fehlen) von Interesse sind. Darüber hinaus werden in dem Fall, in dem ein positiver PhyloCSF-Score beobachtet wird, Schritte beschrieben, um das mikroproteinkodierende Potenzial weiter zu analysieren und effizient mehrere Speziesausrichtungen der identifizierten Aminosäuresequenzen zu generieren, um die Sequenzerhaltung zwischen den Spezies zu veranschaulichen. Schließlich werden in der Diskussion mehrere zusätzliche öffentlich zugängliche Ressourcen und Werkzeuge vorgestellt, um identifizierte Mikroproteineigenschaften zu untersuchen, einschließlich vorhergesagter Domänenstrukturen und Einblicke in die mutmaßliche Mikroproteinfunktion.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Das unten beschriebene Protokoll beschreibt die Schritte zum Laden und Navigieren in den PhyloCSF-Browserspuren im UCSC Genome Browser (generiert von Mudge et al.49). Für allgemeine Fragen zum UCSC Genome Browser finden Sie hier ein umfangreiches Genome Browser User's Guide: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Laden des PhyloCSF Track Hubs in den UCSC Genome Browser
- Öffnen Sie ein Internetbrowserfenster und navigieren Sie zum UCSC Genome Browser (https://genome.ucsc.edu/).
- Wählen Sie unter der Überschrift Unsere Tools die Option Hubs verfolgen aus.
HINWEIS: Die Option Track Hubs finden Sie auch auf der Registerkarte Meine Daten . - Geben Sie auf der Registerkarte Öffentliche Hubs PhyloCSF in das Feld Suchbegriffe ein. Klicken Sie auf die Schaltfläche Öffentliche Hubs durchsuchen .
- Verbinden Sie sich mit PhyloCSF, indem Sie auf die Schaltfläche Verbinden für den Hub-Namen PhyloCSF klicken (Beschreibung: Evolutionäres proteinkodierendes Potenzial, gemessen von PhyloCSF ).
HINWEIS: Dieser Track Hub wird auf zahlreiche Baugruppen geladen, einschließlich Mensch (hg19 und hg38) und Maus (mm10 und mm39). - Nachdem Sie auf Verbinden geklickt haben, warten Sie, bis Sie zur UCSC Genome Browser Gateway-Seite (https://genome.ucsc.edu/cgi-bin/hgGateway) weitergeleitet werden.
2. Navigieren zu interessanten Genen mithilfe von Gen-Identifikatoren
- Wählen Sie die abzufragende Art und Genomassemblierung aus. Um eine andere Art (z. B. Maus) abzufragen, wählen Sie die gewünschte Art unter der Überschrift " Arten durchsuchen/auswählen" aus, indem Sie auf das entsprechende Symbol klicken, oder geben Sie die Art in das Textfeld " Art, allgemeinen Namen oder Assembly-ID eingeben" ein.
HINWEIS: Die Baugruppe wird direkt unter der Überschrift Position suchen aufgelistet. In der Regel ist der Standardwert die menschliche Versammlung (z. B. Dez. 2009 [GRCh37/hg19]). - Wählen Sie die zu durchsuchende Baugruppe unter der Überschrift Position suchen mithilfe des Dropdown-Menüs aus.
- Geben Sie die Position, das Gensymbol oder die Suchbegriffe in das Feld Position/Suchbegriff ein und klicken Sie auf Gehe zu, um zu einem Gen von Interesse im Genome Browser zu navigieren.
- Wenn die Suche zu mehreren Übereinstimmungen geführt hat, warten Sie, bis Sie auf eine Seite umgeleitet werden, die die Auswahl einer interessanten Position erfordert. Klicken Sie auf das entsprechende Gen von Interesse.
3. Navigieren zu genomischen Regionen von Interesse mithilfe von Sequenzinformationen
- Navigieren Sie zum UCSC Genome Browser (https://genome.ucsc.edu/) und wählen Sie das BLAST-Like Alignment Tool (BLAT) unter der Überschrift Unsere Werkzeuge , um eine bestimmte DNA- oder Proteinsequenz abzufragen. Alternativ können Sie den Mauszeiger über die Registerkarte Extras bewegen und die Option Blat auswählen oder diesem Link folgen: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Wählen Sie die Spezies (Genom) und die Zusammenstellung von Interesse über die Dropdown-Menüs aus.
- Definieren Sie den Abfragetyp über das Dropdown-Menü.
- Fügen Sie die gewünschte Sequenz in das Textfeld BLAT Search Genome ein und klicken Sie auf Senden.
- Klicken Sie auf den Browser-Link unter der Überschrift AKTIONEN , um zu der genomischen Region von Interesse zu navigieren.
4. Identifizierung konservierter sORFs mithilfe von PhyloCSF-Track-Daten
- Scannen Sie den genomischen Interessenbereich visuell auf eine positive Bewertung der PhyloCSF-Regionen (Abbildung 1).
HINWEIS: Eine detaillierte Erklärung, wie PhyloCSF-Scores im UCSC Genome Browser visuell interpretiert werden können, finden Sie im Abschnitt "Repräsentative Ergebnisse" weiter unten. - Verwenden Sie die Zoomfunktion, um Bereiche von Interesse zu vergrößern, um Sequenzmerkmale zu untersuchen und nach Start/Stopp-Codons zu suchen. Um manuell zu zoomen, halten Sie die Umschalttaste gedrückt und klicken und halten Sie die Maustaste gedrückt, während Sie den gewünschten Bereich ziehen. Alternativ können Sie die Schaltflächen "Vergrößern" und "Verkleinern" oben auf der Seite verwenden, um zu navigieren (Optionen für den 1,5-fachen, 3-fachen, 10-fachen oder Basiszoom sind verfügbar).
HINWEIS: Bevor Sie die Schaltflächen zum Vergrößern / Verkleinern verwenden , ist es notwendig, das Gen so neu zu positionieren, dass sich der interessierende Bereich in der Mitte des Bildschirms befindet. Um diese Aktion auszuführen, klicken Sie auf das Bild und ziehen Sie es nach links oder rechts, um die genomische Region wie gewünscht horizontal zu verschieben, oder verwenden Sie die Verschiebepfeile oben auf der Seite. - Vergrößern Sie, bis die Nukleotidsequenz (Basensequenz) sichtbar ist.
HINWEIS: Die Nukleotidsequenz erscheint direkt über dem +1 Smoothed PhyloCSF-Score. - Scannen Sie visuell die Nukleotidsequenz am Anfang und Ende der positiv bewerteten PhyloCSF-Regionen, um mutmaßliche Start- (ATG) und Stopp-Codons (TGA/TAA/TAG) zu identifizieren.
HINWEIS: Wenn sich das interessierende Gen auf dem Minusstrang der DNA befindet, sind die Start- und Stopp-Codons das umgekehrte Komplement (dh CAT für das Start-Codon und TCA / TTA / CTA für das Stop-Codon).
5. Homologe Regionen in anderen Genomen betrachten
- Bewegen Sie den Mauszeiger über die Überschrift Ansicht oben auf der Seite und klicken Sie auf die Option In anderen Genomen (Konvertieren).
- Definieren Sie das Genom von Interesse über das Dropdown-Menü unter der Überschrift Neues Genom.
- Wählen Sie im Dropdown-Menü unter der Überschrift Neue Assemblierung die gewünschte genomische Assemblierung aus und klicken Sie dann auf die Schaltfläche Senden .
- Sobald der Browser eine Liste der Bereiche in der neuen Assembly mit Ähnlichkeit zurückgibt, klicken Sie auf den Link zur Chromosomenposition , um zur homologen Region von Interesse zu navigieren.
HINWEIS: Der Prozentsatz der Gesamtbasen (Nukleotide) und die Spanne, die von der Region abgedeckt werden, werden für jede aufgeführte Region definiert. Je höher der Prozentsatz der übereinstimmenden Basen ist, desto höher ist die Erhaltung für die Region von Interesse. - Befolgen Sie die gleichen Navigationsstrategien, die in Abschnitt 4 beschrieben sind, um die Sequenz zu analysieren.
6. Generierung von Multispezies-Sequenzausrichtungen für Mikroproteine von Interesse
- Klicken Sie auf das Gen von Interesse in der GENCODE-Spur im UCSC Genome Browser (in Abbildung 1A mit einem blauen Kästchen dargestellt), um zur Genbeschreibungsseite zu navigieren.
- Klicken Sie unter der Überschrift Sequenz und Links zu Tools und Datenbanken auf den Link in der Tabelle, der die FASTA anderer Arten enthält.
- Klicken Sie auf die Kästchen, die mit den interessierenden Arten verknüpft sind, um sie auszuwählen. Klicken Sie auf Senden. Kopieren Sie die Sequenzen, die unten auf der Seite im FASTA-Format angezeigt werden, und fügen Sie sie in ein Textverarbeitungsdokument ein.
- Öffnen Sie ein zweites Browserfenster und navigieren Sie zum Clustal Omega Multiple Sequence Alignment Tool52 auf der Website53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/ des European Bioinformatics Institute (EMBL-EBI).
- Fügen Sie die Sequenzdateien, die sich noch in der Zwischenablage befinden, in das Feld in SCHRITT 1 ein, das Sequenzen in einem beliebigen unterstützten Format liest. Scrollen Sie zum Ende der Seite und klicken Sie auf Senden. Unter den ausgerichteten Ergebnissen (in schwarzer Schrift) finden Sie Symbole, die den Erhaltungsgrad jeder Aminosäure angeben (Symbole sind in Tabelle 1 definiert).
HINWEIS: Es kann einige Minuten dauern, bis die Ausrichtung generiert ist. - Um die Aminosäureeigenschaften in Farbe anzuzeigen, klicken Sie auf den Link Farben anzeigen direkt über den Sequenzen, um die Aminosäuren entsprechend ihren Eigenschaften zu färben (definiert in Tabelle 2).
- Kopieren Sie die Sequenzausrichtung und fügen Sie sie in ein Textverarbeitungs- oder Diashowprogramm ein, um eine Abbildungs- oder Illustrationsdatei zu generieren (z. B. Abbildung 2).
HINWEIS: Verwenden Sie für die Ausrichtung eine Schriftart mit monospaced, z. B. Courier. - Um andere Ausgaben von der Clustal Omega-Ergebnisseite anzuzeigen, klicken Sie auf die entsprechenden Registerkarten (z. B. Guide Tree oder Phylogenetic Tree).
- Klicken Sie auf die Registerkarte Ergebnisanzeiger , um Optionen zum Anzeigen der Sequenzinformationen mit Jalview anzuzeigen, einem kostenlosen Programm, das sich auf die Bearbeitung, Visualisierung und Analyse mehrerer Sequenzen spezialisiert hat55, oder um auf direkte Links zu MView und Simple Phylogeny56 zuzugreifen.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Hier zeigen wir am Beispiel des validierten Mikroproteins Mitoregulin (Mtln), wie ein konservierter sORF einen positiven PhyloCSF-Score generiert, der im UCSC Genome Browser leicht visualisiert und analysiert werden kann. Mitoregulin wurde zuvor als nicht-kodierende RNA annotiert (früher humane Gen-ID LINC00116 und Maus-Gen-ID 1500011K16Rik). Vergleichende Methoden der Genomik und der Sequenzerhaltungsanalyse spielten eine entscheidende Rolle bei der ersten Entdeckung 40,57,58,59,60,61, was die Stärke dieser Methoden unterstreicht. Für dieses Beispiel wird die Maus GRCm38/mm10 (Dez. 2011) verwendet. Die Suche kann unter Verwendung der Gen-Identifikatoren (Mitoregulin, Mtln) oder der Genposition (chr2:127,791,364-127,792,496) durchgeführt werden, wie in Protokollabschnitt 2 beschrieben. Alternativ kann die Aminosäuresequenz für Mitoregulin (siehe Abbildung 2) mit dem BAT-Tool (beschrieben in Protokollabschnitt 3) durchsucht werden.
Ein Bildschirm ähnlich dem in Abbildung 1A dargestellten wird mit dem PhyloCSF Track Hub angezeigt, der oben auf dem Bildschirm sichtbar ist. Die geglätteten PhyloCSF-Spuren (geglättet mit einem versteckten Markov-Modell, das eine Wahrscheinlichkeit definiert, dass jedes Codon codiert) werden als sechs Gesamtspuren dargestellt, wobei drei Spuren dem Plusstrang der DNA entsprechen (grün dargestellt als PhyloCSF +1, +2 und +3) und drei Spuren dem Minusstrang der DNA entsprechen (rot dargestellt als PhyloCSF -1, -2 und -3). Diese Spuren stellen die drei potenziellen Leserahmen für das Gen von Interesse in jeder Richtung dar. Im Browserfenster werden Exons als blaue Rechtecke dargestellt, die durch dünne blaue horizontale Linien verbunden sind, die die Introns darstellen. Die Pfeilspitzen auf den intronischen Regionen geben an, in welche Richtung das Gen transkribiert wird (und somit auf welchen Strang man sich für den PhyloCSF-Score konzentrieren sollte). Für das Beispiel von Mtln in Abbildung 1 zeigen die intronic-Pfeilspitzen nach links. Daher wird das Mtln-Gen aus dem Minusstrang der DNA transkribiert, und der relevante PhyloCSF-Score wird in den Spuren -1, -2 und -3 (in rot) dargestellt.
Jede PhyloCSF-Spur wird als dünne schwarze Linie mit negativen Bewertungsbereichen dargestellt, die hellgrün/rot unter der Linie dargestellt sind, und positiven Bewertungsbereichen, die in dunkelgrün/rot über der Linie angezeigt werden. Wie in der Einleitung beschrieben, zeigt ein positiver PhyloCSF-Score eine konservierte Region an, die wahrscheinlich codiert. Beachten Sie, dass sie für proteinkodierende Regionen mit besonders hoher Sequenzerhaltung oft auch auf dem Antisense-Strang positiv abschneiden; Der PhyloCSF-Score ist jedoch in der Regel auf dem richtigen Strang höher. Dies ist beispielsweise in Abbildung 1 für Mtln zu sehen, wo die korrekte Kodierungssequenz in der PhyloCSF-1-Spur sehr hoch bewertet wird und der Antisense-Strang (PhyloCSF +2-Spur) ebenfalls eine positive Bewertung generiert. Wie in Abbildung 1A (mit Black Box gekennzeichnet) zu sehen ist, gibt es im ersten Exon von Mtln eine Region, die auf der PhyloCSF-1-Spur sehr gut abschneidet, was darauf hindeutet, dass dies einer Codierungsregion entsprechen könnte. Um diesen Bereich genauer zu untersuchen, ist es hilfreich, den Bereich zu vergrößern und zu vergrößern (Abbildung 1B). Wie in Abbildung 1C,D gezeigt, beginnt der positiv bewertete Bereich im ersten Exon von Mtln direkt über einem Start-Codon (Abbildung 1C) und endet an einem Stopp-Codon (Abbildung 1D), was darauf hindeutet, dass dieser ORF stark konserviert ist und stark darauf hindeutet, dass es sich um einen kodierenden ORF handelt. Da sich Mtln auf dem Minusstrang der DNA befindet, werden die Start- und Stopp-Codons als umgekehrtes Komplement des Codons dargestellt (d. h. das ATG-Startcodon wird als CAT [Abbildung 1C] und das TGA-Stopp-Codon als TCA [Abbildung 1D] dargestellt).
Neben der Verwendung von PhyloCSF zur Suche nach konservierten Regionen mit mikroproteinkodierendem Potenzial kann diese Technik auch als First-Pass-Analyse von mutmaßlichen nicht-kodierenden RNAs angewendet werden, um das Vorhandensein eines konservierten ORF auszuschließen und so eine nicht-kodierende Annotation zu unterstützen. Zum Beispiel zeigt die Analyse der gut charakterisierten lncRNA HOTAIR62,63 mit PhyloCSF einen negativen Score über das gesamte Gen über alle sechs Spuren (Abbildung 3), was stark auf einen Mangel an Sequenzerhaltung hinweist und unterstützt, dass HOTAIR korrekt als nicht-kodierende RNA annotiert ist.
Wie in Abbildung 1 deutlich zu sehen ist, befindet sich der gesamte kodierende ORF für Mitoregulin innerhalb eines einzigen Exons, wodurch eine einfache und unkomplizierte Auslesung durch PhyloCSF mit einer einzigen, ununterbrochenen, positiv bewerteten Region erzeugt wird. PhyloCSF-Track-Hub-Daten sind jedoch nicht immer so klar und einfach zu interpretieren. Zum Beispiel zeigt das Mitolamban / Stmp1 / Mm47-Mikroprotein, das vom Maus-1810058I24Rik-Gen 47,64,65 kodiert wird, einen konservierten ORF, der sich über drei Exons erstreckt (Abbildung 4A), und der positive PhyloCSF-Score springt von der +2-Spur in Exon 1 (Abbildung 4B) zur +3-Spur in Exon 2 (Abbildung 4C) und dann zurück zur +2-Spur in Exon 3 (Abbildung 4D) ). Während dies auf den ersten Blick verwirrend aussieht, ist die Erklärung ziemlich einfach. PhyloCSF bewertet die sechs potenziellen Leserahmen (drei auf dem Plusstrang der DNA und drei auf dem Minusstrang) genomischer Regionen, ohne die spezifische Exon/Intron-Architektur für jedes Gen zu berücksichtigen. Daher behält es die intronische Sequenzinformation in der 3-Nukleotid-Periodizität der Leserahmen bei. Wenn also ein Intron eine Anzahl von Nukleotiden enthält, die nicht durch drei teilbar ist (dh drei Nukleotide / Codon), springt der PhyloCSF-Leserahmen von einer Spur zur anderen.
Schließlich kann PhyloCSF auch effektiv verwendet werden, um mehrere verschiedene kodierende ORFs innerhalb eines einzigen RNA-Moleküls zu identifizieren. Zum Beispiel ist das MIEF1-Mikroprotein (MIEF1-MP) innerhalb der 5'-UTR des mitochondrialen Dehnungsfaktors 1 (MIEF1)66 kodiert (Abbildung 5). Wenn die MIEF1-Genomregion von PhyloCSF analysiert wird, kann ein diskreter positiver PhyloCSF-Score, der dem MIEF1-MP entspricht (Abbildung 5C), leicht vor dem Haupt-CDS für MIEF1 beobachtet werden (Abbildung 5B). Weitere Diskussionen über MIEF1 und sein assoziiertes Mikroprotein (MIEF1-MP) werden unten in der Diskussion zusammen mit einer Zusammenfassung der Stärken und Schwächen der in diesem Artikel beschriebenen Methoden und Protokolle bereitgestellt.

Abbildung 1: Die PhyloCSF-Analyse des Mitoregulin-Gens (Mtln) zeigt eine Region mit hoher Sequenzerhaltung, die einem validierten Mikroprotein entspricht. (A) Screenshots des UCSC Genome Browser und PhyloCSF Tracks zeigen, dass Mtln zwei Exons und ein einzelnes Intron enthält. Die Pfeilspitzen innerhalb des Introns zeigen nach links, was darauf hinweist, dass das Mtln-Gen aus dem Minusstrang der DNA transkribiert wurde, und die relevanten PhyloCSF-Scores werden daher in den Spuren -1, -2 und -3 (rot) angezeigt. Die komplette Mitoregulin-Codierungssequenz ist in Exon 1 enthalten und punktet auf der PhyloCSF-1-Spur (B). Ein konserviertes Start-Codon kann deutlich am Anfang des positiv bewerteten Bereichs in der PhyloCSF-1-Spur (C) beobachtet werden, die mit einem grünen Kasten (CAT, umgekehrtes Komplement ATG) hervorgehoben ist. Zusätzlich wird ein konserviertes Stopp-Codon (TCA, Reverse Complement TGA) mit einem roten Kästchen im Panel (D) angezeigt, das mit dem Ende der positiv bewerteten PhyloCSF-Region übereinstimmt. Detaillierte Informationen über das Mtln-Gen finden Sie, indem Sie auf den Mtln-Gen-Identifikator in der blauen Box klicken (siehe Panel A). Bemerkenswert ist, dass hochkonservierte proteinkodierende Regionen oft auch auf dem Antisense-Strang positiv abschneiden (hier im PhyloCSF +2-Track für Mtln). Der PhyloCSF-Score ist jedoch in der Regel auf dem richtigen Strang höher (in diesem Beispiel die PhyloCSF-Spur -1). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.

Abbildung 2: Multiple Spezies-Sequenzausrichtung des Mikroproteins Mitoregulin, das mit dem Clustal Omega-Programm erzeugt wurde. Die Mitoregulin-Aminosäuresequenzen für die acht angegebenen Spezies wurden wie in Protokollabschnitt 6 beschrieben extrahiert und mit dem Clustal Omega Multiple Sequence Alignment-Tool abgestimmt. Die Eigenschaften der Aminosäuren werden durch die Farbe (rot, klein/hydrophob; blau, sauer; Magenta, basisch; grün, Hydroxl/Sulfhydryl/amin) angezeigt (weiter definiert in Tabelle 2). Die Symbole unter den Aminosäuren geben den Grad der Konservierung an (Sternchen, vollständig konservierte Rückstände; Dickdarm, Aminosäuren mit stark ähnlichen Eigenschaften; Perioden, Konservierung zwischen Gruppen von schwach ähnlichen Eigenschaften) (ausführlich in Tabelle 1 detailliert). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
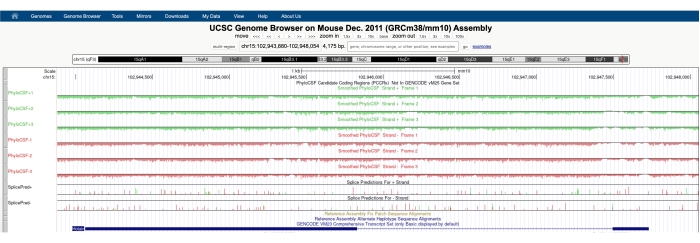
Abbildung 3: Ein Screenshot der PhyloCSF-Spuren für die validierte lange nicht-kodierende RNA Hotair zeigt einen Mangel an Sequenzerhaltung in ihrem gesamten genomischen Locus. Die Pfeilspitzen in der intronischen Region von Hotair zeigen nach links, was darauf hinweist, dass die lncRNA aus dem negativen DNA-Strang transkribiert wird und daher die PhyloCSF-Spuren -1, -2 und -3 im Mittelpunkt der Analyse stehen sollten. Beachten Sie, dass der PhyloCSF-Score im gesamten Gen (für alle sechs Spuren) negativ ist, was auf einen Mangel an Sequenzerhaltung hinweist, was seine korrekte Annotation als nicht-kodierende RNA unterstützt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.

Abbildung 4: PhyloCSF-Analyse des Maus-1810058I24Rik-Gens, das für das Mikroprotein Mitolamban/Stmp1/Mm47 kodiert. (A) Das Maus-1810058I24Rik-Gen besteht aus drei Exons, und die Pfeilspitzen in den intronischen Regionen zeigen nach rechts, was darauf hinweist, dass es auf dem Plusstrang der DNA transkribiert ist und daher die PhyloCSF-Spuren +1, +2 und +3 analysiert werden sollten. Die konservierte Mikroprotein-Kodierungssequenz umfasst alle drei Exons, beginnend mit Exon 1 (B), Lesen durch Exon 2 (C) und endend mit Exon 3 (D). Beachten Sie, dass der positive PhyloCSF-Score auf der +2-Spur in Exon 1, der +3-Spur in Exon 2 und der +2-Spur in Exon 1 zu finden ist. Der Grund für die Bewegung des positiven Scores von einer Spur zur anderen ist, dass PhyloCSF die sechs potenziellen Leserahmen der DNA-Sequenz unabhängig von der Exon/Intron-Struktur des Gens analysiert. Daher führt ein Intron, das eine Anzahl von Nukleotiden enthält, die nicht durch drei (drei Nukleotide / Codon) teilbar sind, zu einer Verschiebung des Leserahmens auf eine andere Spur. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.

Abbildung 5: Die Analyse des genomischen Locus Mief1 mit PhyloCSF identifiziert eine Region mit proteinkodierendem Potenzial in der 5' UTR, die unabhängig vom Haupt-Mief1-CDS auf der gemeinsamen RNA ist. Es wurde gezeigt, dass dieses konservierte vorgeschaltete ORF (uORF) ein Mikroprotein namens Mief1-MP kodiert. (A) Überblick über den genomischen Locus Mief1. Die Pfeilspitzen in den Introns zeigen nach rechts, was darauf hinweist, dass Mief1 aus dem Plusstrang der DNA transkribiert wird (konzentrieren Sie sich auf die PhyloCSF-Spuren +1, +2 und +3, um das Kodierungspotenzial zu bestimmen). Das Haupt-Mief1-CDS kodiert für ein 463-Aminosäuren-Protein und ist in Panel (B) dargestellt. Es gibt jedoch auch einen deutlich konservierten vorgelagerten ORF innerhalb der 5 'UTR von Mief1, der ein einzigartiges 70-Aminosäure-Mikroprotein namens Mief1-MP (C) kodiert. Wie in Panel C zu sehen ist, hat das Mief1-MP sein eigenes konserviertes Start- und Stopp-Codon innerhalb des Mief1 5 'UTR, und der ORF schneidet auf der PhyloCSF + 1-Spur sehr gut ab, was starke Beweise dafür liefert, dass es ein funktionelles Mikroprotein kodiert. Abkürzungen: ORF = offener Leserahmen; uORF = vorgelagerter ORF; UTR = nicht übersetzte Region; CDS = Kodierungssequenz. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
| Symbol | Grad der Aminosäureerhaltung | Gruppierte Aminosäuren |
| Sternchen (*) | Vollständig konservierte Rückstände | Nicht anwendbar (einzelne, vollständig konservierte Rückstände) |
| Doppelpunkt (:) | Gruppen mit stark ähnlichen Eigenschaften | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW |
| Zeitraum (.) | Gruppen mit schwach ähnlichen Eigenschaften | CSA; ATV; SAG; STNK; BBS; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Leerzeichen (kein Symbol) | Keine Ähnlichkeit | Nicht zutreffend (keine Ähnlichkeit) |
Tabelle 1: Definitionen von Konsenssymbolen für Multiple Sequence Alignments, die von Clustal Omega generiert wurden. Die in Abbildung 2 gezeigte Ausrichtung der Sequenz mehrerer Arten wurde mit Clustal Omega52 erzeugt. Abkürzungen: Serin (S), Threonin (T), Alanin (A), Asparagin (N), Glutaminsäure (E), Glutamin (Q), Lysin (K), Asparaginsäure (D), Arginin (R), Methionin (M), Isoleucin (I), Leucin (L), Phenylalanin (F), Histidin (H), Tyrosin (Y), Tryptophan (W), Cystein (C), Valin (V), Glycin (G), Prolin (P).
| Schriftfarbe | Eigentum | Aminosäurerest [Abkürzung] |
| Rot | Klein, hydrophob | Alanin [A], Valin [V], Phenylalanin [F], Prolin [P], Methionin [M], Isoleucin [I], Leucin [L], Tryptophan [W] |
| Blau | Sauer | Asparaginsäure [D], Glutaminsäure [E] |
| Magenta | Grundlegend | Arginin [R], Lysin [K] |
| Grün | Hydroxl, Sulfhydryl, Amin, +G | Serin [S], Threonin [T], Tyrosin [Y], Histidin [H], Cystein [C], Asparagin [N], Glycin [G], Glutamin [Q] |
Tabelle 2: Eigenschaften der in Abbildung 2 dargestellten Aminosäuren. Clustal Omega52 wurde verwendet, um die in Abbildung 2 gezeigte Ausrichtung mehrerer Sequenzen zu erzeugen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Das hier vorgestellte Protokoll enthält detaillierte Anweisungen zur Abfrage genomischer Regionen von Interesse für Mikroprotein-kodierendes Potenzial mit PhyloCSF auf dem benutzerfreundlichen UCSC Genome Browser 48,49,50,51. Wie oben beschrieben, ist PhyloCSF ein leistungsstarker vergleichender Genomik-Algorithmus, der phylogenetische Modelle und Codon-Substitutionsfrequenzen integriert, um evolutionäre Signaturen zu identifizieren, die typisch für proteinkodierende Genesind 48,49. PhyloCSF wurde häufig verwendet, um funktionelle Mikroproteine in genomischen Regionen zu identifizieren, die zuvor als nicht-kodierend annotiert wurden 38,39,40,41,42,43,44,45,46,47 , und es hat sich gezeigt, dass dieser Ansatz andere vergleichende genomische Methoden für kurze Sequenzen wie Mikroproteine, die so klein wie 13 Aminosäuren sind, und für kleine Exons von kanonischen Proteinen 35,48,49 übertrifft. Insbesondere der Nutzen von PhyloCSF als robuste Methode zur Identifizierung funktioneller proteinkodierender Sequenzen durch evolutionäre Konservierung geht über die von Wirbeltieren und wirbellosen Arten hinaus und wurde kürzlich sogar auf virale Genome angewendet, um die proteinkodierende Kapazität des SARS-CoV-2-Genoms67 erfolgreich abzufragen.
Neben der Identifizierung mutmaßlicher kodierender Sequenzen in annotierten nicht-kodierenden RNAs besteht ein Vorteil von PhyloCSF darin, dass es auch konservierte Mikroproteine, die von ORFs kodiert werden, in annotierten untranslatierten Regionen (UTRs) von kanonischen proteinkodierenden Genen zuverlässig nachweisen kann, einschließlich 5' Upstream- und 3' Downstream-ORFs (uORFs bzw. dORFs)8,19,66,68 . Zum Beispiel ist das MIEF1-Mikroprotein (MIEF1-MP) im 5' UTR des mitochondrialen Dehnungsfaktors 1 (MIEF1)66 kodiert. Im Fall von MIEF1-MP wird ein diskreter positiver PhyloCSF-Score, der dem MIEF1-MP entspricht, vor dem ORF beobachtet, der MIEF1 kodiert (Abbildung 5). Während einige uORF-kodierte Mikroproteine direkt mit den nachgeschalteten kanonischen Proteinen auf ihrer gemeinsamen mRNA interagieren (z. B. MIEF1-MP und MIEF1), funktionieren andere unabhängig von dem Protein, das vom Haupt-CDS66,68 kodiert wird. Daher sollte bei der Charakterisierung von uORF-kodierten Mikroproteinen nicht davon ausgegangen werden, dass sie durch direkte Regulation ihres nachgeschalteten Proteinprodukts funktionieren.
Während PhyloCSF viele klare Stärken als Werkzeug zur Identifizierung konservierter Mikroprotein-kodierender Sequenzen hat, ist es wichtig, einige Einschränkungen dieser Methode zu erkennen. Erstens, während die Sequenzkonservierung stark darauf hindeutet, dass eine genomische Region eine funktionelle Selektion durchlaufen hat und somit kodiert, schließen ein Mangel an robuster Konservierung und ein daraus resultierender negativer PhyloCSF-Score das Kodierungspotenzial für eine bestimmte Sequenz nicht definitiv aus. Mit anderen Worten, wenn man sich ausschließlich auf PhyloCSF verlässt, kann dies zur Aufsicht über übersetzte ORFs führen, die nicht stark konserviert sind, aber dennoch funktionelle Mikroproteine produzieren. Insbesondere genomische Regionen mit niedrigen Erhaltungs- oder negativen Erhaltungswerten könnten artspezifischen kodierenden Regionen oder denen evolutionärer "junger" Gene über Sequenzdivergenz oder De-novo-Gengeburt 46,69,70,71,72,73,74 entsprechen. Zum Beispiel wird das Mikroprotein ASAP, das von der ehemaligen nicht-kodierenden menschlichen RNA LINC00467 kodiert wird, von PhyloCSF nicht positiv bewertet, da die Aminosäuresequenz nur bei höheren Säugetieren75 konserviert wird. Darüber hinaus identifizierten neuere Studien mehrere humanspezifische Mikroproteine, darunter eines, das von der intergenen lncRNA RP3-527G5.1 kodiert wird, das keinen positiven PhyloCSF-Score68,72 erzeugt. In dieser Hinsicht kann das Fehlen eines positiven PhyloCSF-Scores nicht als Beweis für eine nichtkodierende Region interpretiert werden und sollte mit Vorsicht interpretiert werden.
Eine zweite Überlegung, die bei der Verwendung von PhyloCSF zu beachten ist, ist, dass, obwohl ein positiver Score stark auf funktionelle Selektion und Proteinkodierungskapazität hindeutet, diese Beweislinie nicht allein stehen kann und experimentell validiert werden muss. Beispiele für Methoden, die verwendet werden können, um unterstützende Beweise für eine stabile Mikroproteinexpression zu generieren, sind der Nachweis des mutmaßlichen Proteins durch Massenspektrometrie oder Western Blotting unter Verwendung eines Antikörpers, der gegen die interessierende Mikroproteinsequenz erhoben wird. Da es aufgrund des Mangels an Sequenzmöglichkeiten für eine optimale Antigenität schwierig sein kann, zuverlässige Antikörper für Mikroproteine zu erzeugen, ist es alternativ auch möglich, CRISPR/Cas9 und den homologiegesteuerten Reparaturweg (HDR) zu verwenden, um einen Epitop-Tag in den endogenen Locus im Rahmen der mutmaßlichen Mikroproteinsequenz einzuführen und so den Nachweis des interessierenden Proteins mit einem hochaffinen Antikörper (z. B. FLAGGE, HA, V5, Myc)18. Eine letzte Einschränkung von PhyloCSF ist, dass, obwohl es derzeit in viele der häufig verwendeten genomischen Anordnungen integriert ist, einschließlich Homo sapiens (human hg19, hg38), Mus musculus (Maus mm10, mm39), Gallus gallus (Huhn, galGal4, galGal6), Drosophila melanogaster (Fruchtfliege, dm6), Caenorhabditis elegans (Nematoden, ce11) und SARS-CoV-2 (wuhCor1) gibt es noch viele Arten, die derzeit nicht direkt im UCSC Genome Browser abgefragt werden können.
Die Identifizierung konservierter Domänen oder Sequenzmerkmale innerhalb identifizierter Mikroproteine kann dazu beitragen, das Vertrauen in ihre funktionelle Relevanz zu erhöhen und einen Einblick in ihre mutmaßliche Funktion zu geben. Hier geben wir Empfehlungen für spezifische Werkzeuge und Ressourcen, mit denen identifizierte Mikroprotein-Aminosäuresequenzen genauer analysiert werden können, um solche Erkenntnisse zu gewinnen. Die unten aufgeführten (und in der Materialtabelle zusammengefassten) spezifischen Werkzeuge sind für die Öffentlichkeit frei zugänglich, und wir haben sie in Mikroproteinstudien 18,38,39,40,41,47 als besonders benutzerfreundlich und robust empfunden. Über die hier beschriebenen Tools hinaus gibt es eine Vielzahl zusätzlicher Ressourcen, die in bioinformatischen Ressourcenportalen wie Expasy (https://www.expasy.org) und EMBL-EBI (https://www.ebi.ac.uk/services/all) zu finden sind. Die Einzelheiten der einzelnen Tools in diesen Repositories gehen jedoch über den Rahmen dieses Artikels hinaus. Hier empfehlen wir die folgenden Ressourcen.
Zunächst analysiert TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) Proteinsequenzen von Interesse auf das Vorhandensein von Transmembrandomänen. Bemerkenswert ist, dass eine Reihe von Mikroproteinen, die bisher funktionell charakterisiert wurden, Single-Pass-Transmembrandomänen enthalten, was ihre Lokalisierung in Membranregionen erleichtert und ihre direkte Regulation von Ionenkanälen, Austauschern und membranassoziierten Enzymenermöglicht 30. Zweitens ist das National Center for Biotechnology Information (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) ein beliebtes Werkzeug, um konservierte Domänen innerhalb von Protein- oder kodierenden Nukleotidsequenzen zu identifizieren. Drittens bietet die Proteinfamilie (Pfam)78-Datenbank (http://pfam.xfam.org) Ausrichtungen und Klassifikationen von Proteinfamilien und -domänen. Viertens ist WoLF PSORT79 (https://wolfpsort.hgc.jp/) ein Werkzeug, mit dem die subzelluläre Proteinlokalisation vorhergesagt werden kann. Fünftens ist COXPRESdB80 eine Gen-Coexpressions-Datenbank (https://coxpresdb.jp), die koregulierte Genbeziehungen zur Abschätzung von Genfunktionen bereitstellt. Schließlich ist SignalP 6.081 ein weit verbreitetes Vorhersageprogramm (https://services.healthtech.dtu.dk/service.php?SignalP), das das Vorhandensein einer Signalpeptidsequenz erkennt und die Position der Spaltungsstelle vorhersagt.
Zusammenfassend lässt sich sagen, dass die hier beschriebenen Methoden verwendet werden können, um genomische Regionen von Interesse auf Proteinkodierungspotenzial mit PhyloCSF im UCSC Genome Browser effektiv zu analysieren. Diese Methoden sind sehr zugänglich und können von Einzelpersonen ohne vorherige Ausbildung oder Fachwissen in Bioinformatik oder vergleichender Genomik leicht erlernt und effizient angewendet werden. Wie hier im Detail gezeigt, ist PhyloCSF ein leistungsfähiges Werkzeug, das als First-Pass-Analyse angewendet werden kann, um proteinkodierende von nicht-kodierenden Genen in Wirbeltier-, Wirbellosen- und Virusgenomen zu unterscheiden, und die Stärken dieses Ansatzes überwiegen bei weitem die festgestellten Schwächen.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Acknowledgments
Diese Arbeit wurde durch Zuschüsse der National Institutes of Health (HL-141630 und HL-160569) und der Cincinnati Children's Research Foundation (Trustee Award) unterstützt.
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
