

Summary
O protocolo descrito aqui fornece instruções detalhadas sobre como analisar regiões genômicas de interesse para potencial de codificação de microproteínas usando PhyloCSF no Navegador de Genoma UCSC fácil de usar. Além disso, várias ferramentas e recursos são recomendados para investigar mais as características sequenciais das microproteínas identificadas para obter informações sobre suas funções putativas.
Abstract
O sequenciamento de última geração (NGS) impulsionou o campo da genômica para a frente e produziu sequências de genomas inteiros para inúmeras espécies animais e organismos modelo. No entanto, apesar dessa riqueza de informações sequenciais, esforços abrangentes de anotação genética têm se mostrado desafiadores, especialmente para pequenas proteínas. Notavelmente, os métodos convencionais de anotação de proteínas foram projetados para excluir intencionalmente proteínas putativas codificadas por quadros de leitura aberto curtos (sORFs) com menos de 300 nucleotídeos de comprimento para filtrar o número exponencialmente maior de sORFs não codificadores espúrios em todo o genoma. Como resultado, centenas de pequenas proteínas funcionais chamadas microproteínas (<100 aminoácidos em comprimento) foram incorretamente classificadas como RNAs não codificantes ou negligenciadas inteiramente.
Aqui fornecemos um protocolo detalhado para aproveitar ferramentas bioinformáticas gratuitas e disponíveis publicamente para consultar regiões genômicas para potencial de codificação de microproteínas com base na conservação evolutiva. Especificamente, fornecemos instruções passo a passo sobre como examinar o potencial de conservação e codificação de sequências usando as Frequências de Substituição de Codon Filogenéticas (PhyloCSF) no navegador de genoma da Universidade da Califórnia Santa Cruz (UCSC). Além disso, detalhamos etapas para gerar eficientemente alinhamentos de várias espécies de sequências de microproteínas identificadas para visualizar a conservação da sequência de aminoácidos e recomendar recursos para analisar características de microproteínas, incluindo estruturas de domínio previstas. Essas ferramentas poderosas podem ser usadas para ajudar a identificar sequências putativas de codificação de microproteínas em regiões genômicas não anônicas ou para excluir a presença de uma sequência de codificação conservada com potencial translacional em uma transcrição de interesse não codificada.
Introduction
A identificação do conjunto completo de elementos de codificação no genoma tem sido um objetivo importante desde o início do Projeto Genoma Humano, e continua sendo um objetivo central para a compreensão dos sistemas biológicos e da etiologia das doenças de base genética 1,2,3,4. Os avanços nas técnicas de NGS levaram à produção de sequências de genomas inteiras para um grande número de organismos, incluindo vertebrados, invertebrados, leveduras e plantas5. Além disso, métodos de sequenciamento transcricional de alto rendimento revelaram ainda a complexidade do transcriptome celular, e identificaram milhares de novas moléculas de RNA com funções de codificação de proteínas e não codificação 6,7. A decodificação dessa vasta quantidade de informações sequenciais é um processo contínuo, e os desafios permanecem com esforços abrangentes de anotação genética8.
O recente desenvolvimento de métodos de criação de perfil translacional, incluindo o perfil ribossomo 9,10 e o sequenciamento poli-ribossomo11, forneceram evidências indicando que centenas de eventos de tradução nãocanônica mapeiam para sORFs não anotados atualmente em todo o genoma, com potencial para gerar pequenas proteínas chamadas microproteínas ou micropeptídeos12,13, 14,15,16, 17. As microproteínas emergiram como uma nova classe de proteínas versáteis anteriormente negligenciadas por métodos de anotação genética padrão devido ao seu pequeno tamanho (<100 aminoácidos) e à falta de características genéticas clássicas de codificação de proteínas 8,12,18,19,20. Microproteínas foram descritas em praticamente todos os organismos, incluindo levedura21,22, moscas 17,23,24 e mamíferos 25,26,27,28, e têm mostrado desempenhar papéis críticos em diversos processos, incluindo desenvolvimento, metabolismo e sinalização de estresse 19,20,29, 30,31,32,33,34. Assim, é imperativo continuar a minerar o genoma para membros adicionais desta classe há muito negligenciada de pequenas proteínas funcionais.
Apesar do reconhecimento generalizado da importância biológica das microproteínas, essa classe de genes permanece muito sub-representada nas anotações do genoma, e sua identificação precisa continua sendo um desafio contínuo que tem dificultado o progresso no campo. Várias ferramentas computacionais e métodos experimentais foram recentemente desenvolvidos para superar as dificuldades associadas à identificação de sequências de codificação de microproteínas (discutidas extensivamente em várias revisões abrangentes 8,35,36,37). Muitos estudos recentes de identificação de microproteínas 38,39,40,41,42,43,44,45,46,47 têm confiado fortemente no uso de um algoritmo chamado PhyloCSF 48,49 , uma poderosa abordagem comparativa de genômica que pode ser aproveitada para distinguir regiões conservadas de codificação de proteínas do genoma daquelas que não são codificadas.
O PhyloCSF compara as frequências de substituição de codon (CSF) usando alinhamentos de nucleotídeos de várias espécies e modelos filogenéticos para detectar assinaturas evolutivas de genes codificadores de proteínas. Esta abordagem empírica baseada em modelos baseia-se na premissa de que as proteínas são conservadas principalmente no nível de aminoácidos em vez da sequência de nucleotídeos. Portanto, substituições sinônimos de codon, que codificam o mesmo aminoácido, ou substituições de codon a aminoácidos com propriedades conservadas (ou seja, carga, hidroofobidade, polaridade) são pontuadas positivamente, enquanto substituições não-sinônimos, incluindo substituições missense e sem sentido, pontuam negativamente. O PhyloCSF é treinado em dados de genoma inteiro e provou ser eficaz na pontuação de porções curtas de uma sequência de codificação (CDS) isoladamente da sequência completa, o que é necessário ao analisar microproteínas ou exons individuais de genes padrão de codificação de proteínas48,49.
Notavelmente, a recente integração dos hubs de trilha PhyloCSF no Navegador de Genoma 49,50,51 da Universidade da Califórnia Santa Cruz (UCSC) permite que pesquisadores de todas as origens acessem facilmente uma interface fácil de usar para consultar regiões genômicas de interesse para o potencial de codificação de proteínas. O protocolo descrito abaixo fornece instruções detalhadas sobre como carregar os hubs de rastrear PhyloCSF no Navegador de Genoma da UCSC e, posteriormente, interrogar regiões genômicas de interesse para sondar regiões de codificação de proteínas de alta confiança (ou a falta delas). Além disso, no caso em que um escore phyloCSF positivo é observado, as etapas são delineadas para analisar melhor o potencial de codificação de microproteínas e gerar eficientemente alinhamentos de várias espécies das sequências de aminoácidos identificadas para ilustrar a conservação da sequência de espécies cruzadas. Por fim, vários recursos e ferramentas adicionais disponíveis ao público são introduzidos na discussão para levantamento de características identificadas de microproteínas, incluindo estruturas de domínio previstas e insights sobre a função putativa de microproteínas.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
O protocolo descrito abaixo detalha as etapas para carregar e navegar nas faixas do navegador PhyloCSF no Navegador genoma UCSC (gerado por Mudge et al.49). Para dúvidas gerais sobre o Navegador de Genoma da UCSC, um extenso Guia do Usuário do Navegador genoma pode ser encontrado aqui: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Carregando o PhyloCSF Track Hub para o navegador de genoma UCSC
- Abra uma janela do navegador de internet e navegue até o Navegador de Genoma do UCSC (https://genome.ucsc.edu/).
- Sob o título De ferramentas , selecione a opção Track Hubs .
NOTA: A opção Track Hubs também pode ser encontrada na guia Meus dados . - Na guia Centros Públicos , digite PhyloCSF na caixa de termos 'Pesquisa' . Clique no botão 'Pesquisar Centros públicos' .
- Conecte-se ao PhyloCSF clicando no botão Conectar para o Nome do Hub PhyloCSF (Descrição: potencial de codificação de proteínas evolutivas medida pelo PhyloCSF).
NOTA: Este Track Hub será carregado para numerosos conjuntos, incluindo humano (hg19 e hg38) e mouse (mm10 e mm39). - Depois de clicar em conectar, aguarde ser redirecionado para a página do Gateway do Navegador de Genoma do UCSC (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Navegar para genes de interesse usando identificadores genéticos
- Selecione a espécie e o conjunto do genoma para consultar. Para consultar uma espécie diferente (por exemplo, mouse), selecione a espécie de interesse sob o título Procurar/Selecionar Espécies clicando no ícone apropriado ou digitar a espécie na caixa de texto que diz: Digite espécie, nome comum ou ID de montagem.
NOTA: O conjunto está listado diretamente no título "Encontrar posição ". Normalmente, o padrão é a Assembleia Humana (por exemplo, dez de 2009 [GRCh37/hg19]). - Escolha o conjunto para pesquisar no título 'Encontrar posição' usando o menu suspenso.
- Digite a posição, o símbolo genético ou os termos de pesquisa na caixa "Posição/Termo de Pesquisa " e clique em Ir para navegar até um gene de interesse no Navegador genoma.
- Se a pesquisa resultou em várias correspondências, aguarde ser redirecionada para uma página que exija a seleção de uma posição de interesse. Clique no gene de interesse apropriado.
3. Navegar para regiões genômicas de interesse usando informações de sequência
- Navegue até o Navegador de Genoma do UCSC (https://genome.ucsc.edu/) e selecione a Ferramenta de Alinhamento semelhante a BLAST (BLAT) sob as nossas ferramentas que se dirigem para consultar uma sequência específica de DNA ou proteína. Como alternativa, passe o cursor sobre a guia Ferramentas e selecione a opção Blat ou siga este link: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Selecione as espécies (Genoma) e a Montagem de interesse usando os menus suspensos.
- Defina o tipo Desaída usando o menu suspenso.
- Cole a sequência de interesse na caixa de texto BLAT Search Genoma e clique em Enviar.
- Clique no link do navegador sob o título ACTIONS para navegar até a região genômica de interesse.
4. Identificando sORFs conservados usando dados de faixa PhyloCSF
- Escaneie visualmente a área genômica de interesse para pontuar positivamente as regiões de PhyloCSF (Figura 1).
NOTA: Para obter uma explicação detalhada de como interpretar visualmente os escores do PhyloCSF no Navegador de Genoma do UCSC, consulte a seção de resultados representativos abaixo. - Use o recurso zoom para ampliar regiões de interesse para examinar características de sequência e procurar códons de início/parada. Para ampliar manualmente, segure a tecla shift e clique e segure o botão do mouse enquanto arrasta ao longo da região de interesse. Alternativamente, use os botões de zoom e zoom na parte superior da página para navegar (1,5x, 3x, 10x ou opções de zoom base estão disponíveis).
NOTA: Antes de usar os botões de zoom-in/zoom , é necessário reposicionar o gene para que a região de interesse esteja no meio da tela. Para realizar esta ação, clique na imagem e arraste-a para a esquerda ou para a direita para mover a região genômica horizontalmente conforme desejado ou use as setas de movimento na parte superior da página. - Aproxime-se até que a sequência nucleotídeo (base) esteja visível.
NOTA: A sequência de nucleotídeos aparecerá diretamente acima da pontuação de PhyloCSF suavizada. - Escaneie visualmente a sequência de nucleotídeos perto do início e fim das regiões phyloCSF de pontuação positiva para identificar códons de início putativo (ATG) e parar (TGA/TAA/TAG).
NOTA: Se o gene de interesse estiver no fio negativo do DNA, os códons de início e parada serão o complemento inverso (ou seja, CAT para o códon inicial e TCA/TTA/CTA para o códon de parada).
5. Visualização de regiões homólogas em outros genomas
- Passe o mouse sobre a posição Exibir na parte superior da página e clique na opção Em Outros Genomas (Converter).
- Defina o genoma de interesse usando o menu suspenso abaixo do título Novo Genoma .
- Selecione o conjunto genômico de interesse usando o menu suspenso no título Nova montagem e clique no botão Enviar .
- Uma vez que o navegador retorne uma lista de regiões do novo conjunto com semelhança, clique no link de posição cromossomómero para navegar até a região homólogoa de interesse.
NOTA: O percentual de bases totais (nucleotídeos) e o vão coberto pela região serão definidos para cada região listada. Quanto maior o percentual de bases correspondentes, maior a conservação para a região de interesse. - Siga as mesmas estratégias de navegação detalhadas na Seção 4 para analisar a sequência.
6. Gerando alinhamentos de sequência de várias espécies para microproteínas de interesse
- Clique no gene de interesse na faixa GENCODE no Navegador de Genoma UCSC (indicado na Figura 1A com uma caixa azul) para navegar até a página de descrição do gene.
- Em título Sequência e Links para Ferramentas e Bancos de Dados , clique no link na tabela que lê Outras Espécies FASTA.
- Clique nas caixas associadas à espécie de interesse para selecioná-las. Clique em Enviar. Copie e cole as sequências que aparecem na parte inferior da página no formato FASTA em um documento de processamento de texto.
- Abra uma segunda janela de navegador e navegue até a ferramenta Clustal Omega Multiple Sequence Alignment 52 no site53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Cole os arquivos de sequência que ainda estão na área de transferência na caixa no PASSO 1 que lê sequências em qualquer formato suportado. Role até a parte inferior da página e clique em Enviar. Veja abaixo os resultados alinhados (na fonte preta) para símbolos que indicam o grau de conservação de cada aminoácido (os símbolos são definidos na Tabela 1).
NOTA: Pode levar vários minutos para gerar o alinhamento. - Para ver as propriedades aminoácidos na cor, clique no link Mostrar cores diretamente acima das sequências para colorir os aminoácidos de acordo com suas propriedades (definidas na Tabela 2).
- Copie e cole o alinhamento de sequência em um programa de processamento de palavras ou slideshow para gerar uma figura ou arquivo de ilustração (por exemplo, Figura 2).
NOTA: Use uma fonte monoespaçada para o alinhamento, como o Courier. - Para ver outras saídas da página de resultados Domô Clustal , clique nas guias apropriadas (ou seja, Árvore Guia ou Árvore Filogenética).
- Clique na guia Resultados Para obter opções para visualizar as informações de sequência usando o Jalview, um programa gratuito especializado em edição, visualização e análise de alinhamento de sequência múltipla55, ou para acessar links diretos para MView e Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Aqui usaremos a mitoregulina de microproteína validada (Mtln) como exemplo para demonstrar como um sORF conservado irá gerar uma pontuação PhyloCSF positiva que pode ser facilmente visualizada e analisada no Navegador de Genoma da UCSC. Mitoregulina foi anteriormente anotada como um RNA não codificado (anteriormente iD gene humano LINC00116 e gene do rato ID 150001K16Rik). Os métodos comparativos de genômica e análise de conservação de sequências desempenharam um papel crítico em sua descoberta inicial 40,57,58,59,60,61, destacando a força desses métodos. Para este exemplo, será utilizado o conjunto grcm38/mm10 (dez 2011). A pesquisa pode ser realizada usando os identificadores genéticos (mitoregulina, Mtln) ou a posição genética (chr2:127.791.364-127.792.496) conforme descrito na seção de protocolo 2. Alternativamente, a sequência de aminoácidos para mitoregulina (mostrada na Figura 2) pode ser pesquisada usando a ferramenta BLAT (descrita na seção de protocolo 3).
Uma tela semelhante à retratada na Figura 1A aparecerá com o PhyloCSF Track Hub visível na parte superior da tela. As faixas PhyloCSF Suavizadas (suavizadas com um modelo Markov oculto definindo uma probabilidade de que cada códonte esteja codificando) são retratadas como seis faixas totais, com três faixas correspondentes ao fio mais do DNA (retratado em verde como PhyloCSF +1, +2 e +3) e três faixas correspondentes ao fio menos de DNA (retratado em vermelho como PhyloCSF -1, -2 e -3). Essas faixas representam os três quadros de leitura potenciais para o gene de interesse em cada direção. Na janela do navegador, os exons são retratados como retângulos azuis conectados por finas linhas horizontais azuis, que representam os introns. As pontas de flecha nas regiões intrônicas indicam em que direção o gene é transcrito (e, portanto, em qual fio se concentrar para a pontuação PhyloCSF). Para o exemplo de Mtln na Figura 1, as pontas de flechas intrônicas estão apontando para a esquerda. Portanto, o gene Mtln é transcrito a partir do fio negativo do DNA, e a pontuação phyloCSF relevante é retratada nas faixas -1, -2 e -3 (em vermelho).
Cada faixa PhyloCSF é retratada como uma linha preta fina com regiões de pontuação negativas retratadas em verde claro/vermelho abaixo da linha e regiões de pontuação positiva indicadas em verde escuro/vermelho acima da linha. Como descrito na introdução, um escore PhyloCSF positivo indica uma região conservada que é provavelmente codificação. Note que para regiões de codificação de proteínas com conservação de sequências particularmente elevadas, muitas vezes elas também pontuam positivamente na cadeia antissamo; no entanto, a pontuação PhyloCSF é geralmente maior no fio correto. Por exemplo, isso pode ser visto na Figura 1 para Mtln, onde a sequência de codificação correta é muito alta na faixa PhyloCSF -1, e a cadeia antissense (faixa PhyloCSF +2) também gera uma pontuação positiva. Como visto na Figura 1A (indicada com caixa preta), há uma região no primeiro exon de Mtln que pontua muito bem na faixa PhyloCSF -1, sugerindo que isso pode corresponder a uma região de codificação. Para examinar essa região com mais detalhes, é útil ampliar e ampliar a região (Figura 1B). Como mostrado na Figura 1C,D, a região de pontuação positiva no primeiro exon de Mtln começa diretamente sobre um códon inicial (Figura 1C) e termina em um códon de parada (Figura 1D), o que indica que este ORF é altamente conservado e sugere fortemente que é um ORF codificador. Como o Mtln está no fio negativo do DNA, os códons de início e parada são mostrados como o complemento inverso do códon (ou seja, o codon inicial atg é mostrado como CAT [Figura 1C] e o codon de parada TGA é mostrado como TCA [Figura 1D]).
Além de usar o PhyloCSF para procurar regiões conservadas com potencial de codificação de microproteínas, essa técnica também pode ser aplicada como uma análise de primeira passagem de RNAs putativas não codificadoras para excluir a presença de um ORF conservado, fornecendo suporte para uma anotação não codificadora. Por exemplo, a análise do bem caracterizado lncRNA HOTAIR62,63 usando PhyloCSF mostra um resultado negativo em todo o gene em todas as seis faixas (Figura 3), indicando fortemente a falta de conservação de sequências e fornecendo suporte de que o HOTAIR é corretamente anotado como um RNA não codificador.
Como claramente visto na Figura 1, toda a codificação ORF para mitoregulina está localizada dentro de um único exon, produzindo assim uma leitura simples e direta por PhyloCSF com uma região única, ininterrupta e positivamente pontuadora. No entanto, os dados do hub de controle PhyloCSF nem sempre são tão claros e fáceis de interpretar. Por exemplo, a microproteína mitolamban/Stmp1/Mm47 codificada pelo gene 1810058I24Rik 47,64,65 retrata um ORF conservado que abrange três exons (Figura 4A), e a pontuação positiva do PhyloCSF salta da faixa +2 no exon 1 (Figura 4B) para a faixa +3 no exon 2 (Figura 4C), e depois volta para a faixa +2 no exon 3 (Figura 4D ). Embora à primeira vista isso pareça confuso, a explicação é bastante simples. PhyloCSF pontua os seis quadros de leitura potenciais (três na vertente mais do DNA e três na cadeia menos) de regiões genômicas sem considerar a arquitetura exon/intron específica para cada gene. Portanto, mantém as informações de sequência intrônica na periodicidade 3-nucleotídeo dos quadros de leitura. Assim, se um intron contiver uma série de nucleotídeos que não são divisíveis por três (ou seja, três nucleotídeos/codon), o quadro de leitura PhyloCSF saltará de uma faixa para outra.
Por último, o PhyloCSF também pode ser usado efetivamente para identificar orfs de codificação distintos múltiplos dentro de uma única molécula de RNA. Por exemplo, a microproteína MIEF1 (MIEF1-MP) está codificada dentro do UTR de 5' do fator de alongamento mitocondrial 1 (MIEF1)66 (Figura 5). Quando a região genômica MIEF1 é analisada por PhyloCSF, um escore PhyloCSF positivo discreto correspondente ao MIEF1-MP (Figura 5C) pode ser facilmente observado a montante do CDS principal para MIEF1 (Figura 5B). Outra discussão sobre o MIEF1 e sua microproteína associada (MIEF1-MP) é fornecida abaixo na discussão, juntamente com um resumo dos pontos fortes e fracos dos métodos e protocolos descritos neste artigo.

Figura 1: A análise phyloCSF do gene mitoregulina (Mtln) indica uma região de conservação de alta sequência correspondente a uma microproteína validada. (A) Capturas de tela do Navegador de Genoma UCSC e faixas PhyloCSF mostram que o Mtln contém dois exons e um único intron. As pontas de flecha dentro do ponto intron à esquerda, indicando que o gene Mtln é transcrito a partir do fio negativo do DNA, e os escores phyloCSF relevantes são, portanto, mostrados nas faixas -1, -2 e -3 (em vermelho). A sequência completa de codificação de mitoregulina está contida no Exon 1 e pontua muito na faixa PhyloCSF -1 (B). Um códon de partida conservado pode ser claramente observado no início da região de pontuação positiva na faixa PhyloCSF -1 (C), que é destacada com uma caixa verde (CAT, complemento reverso ATG). Além disso, um códo de parada conservado (TCA, complemento reverso TGA) é indicado com uma caixa vermelha no painel (D), que se alinha com o final da região PhyloCSF de pontuação positiva. Informações detalhadas sobre o gene Mtln podem ser encontradas clicando no identificador do gene Mtln dentro da caixa azul (mostrado no painel A). Note-se que regiões altamente conservadas de codificação de proteínas muitas vezes também pontuam positivamente na cadeia antissense (vista aqui na faixa PhyloCSF +2 para Mtln). No entanto, a pontuação PhyloCSF é tipicamente maior no fio correto (a faixa PhyloCSF -1 neste exemplo). Clique aqui para ver uma versão maior desta figura.

Figura 2: Alinhamento de sequência de espécies múltiplas da mitoregulina de microproteína gerada usando o programa Ômega Clustal. As sequências de aminoácidos mitoregulina para as oito espécies indicadas foram extraídas como detalhadas na seção de protocolo 6 e alinhadas com a ferramenta de alinhamento de sequência múltipla Clustal Omega. As propriedades dos aminoácidos são indicadas por cor (vermelho, pequeno/hidrofóbico; azul, ácido; magenta, básico; verde, hidroxl/sulfhydryl/amine) (ainda definido na Tabela 2). Os símbolos abaixo dos aminoácidos indicam o grau de conservação (asteriscos, resíduos totalmente conservados; cólons, aminoácidos com propriedades fortemente semelhantes; períodos, conservação entre grupos de propriedades fracamente semelhantes) (detalhado extensivamente na Tabela 1). Clique aqui para ver uma versão maior desta figura.
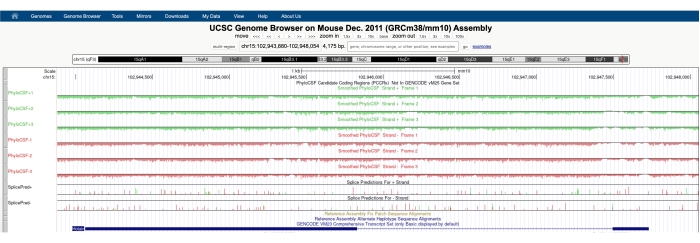
Figura 3: Uma captura de tela das faixas PhyloCSF para o RNA hotair não codificador longo validado mostra uma falta de conservação de sequência em todo o seu lócus genômico. As pontas de flecha na região intrônica de Hotair estão apontando para a esquerda, indicando que o lncRNA é transcrito a partir do fio negativo do DNA, e, portanto, as faixas PhyloCSF -1, -2 e -3 devem ser o foco da análise. Observe que o escore PhyloCSF é negativo em todo o gene (para todas as seis faixas), indicando uma falta de conservação de sequência, que suporta sua anotação adequada como um RNA não codificador. Clique aqui para ver uma versão maior desta figura.

Figura 4: Análise phyloCSF do gene 1810058I24Rik , que codifica a microproteína mitolamban/Stmp1/Mm47. (A) O gene 1810058I24Rik é composto por três exons, e as pontas das flechas nas regiões intrônicas apontam à direita, indicando que ele é transcrito no fio mais do DNA e, portanto, o gene PhyloCSF +1, +2 e +3 faixas devem ser analisados. A sequência de codificação de microproteína conservada abrange todos os três exons, começando em exon 1 (B), lendo através de exon 2 (C), e terminando em exon 3 (D). Note que a pontuação positiva do PhyloCSF é encontrada na faixa +2 no exon 1, na faixa +3 no exon 2 e na faixa +2 no exon 1. A razão para o movimento do resultado positivo de uma faixa para outra é que o PhyloCSF analisa os seis quadros de leitura potenciais da sequência de DNA independente da estrutura exon/intron do gene. Portanto, um intron contendo uma série de nucleotídeos que não são divisíveis por três (três nucleotídeos/codon) causará uma mudança no quadro de leitura para uma faixa diferente. Clique aqui para ver uma versão maior desta figura.

Figura 5: A análise do lócus genômico Mief1 com o PhyloCSF identifica uma região com potencial de codificação de proteínas no UTR de 5' que é independente do principal CDS Mief1 no RNA compartilhado. Este ORF upstream conservado (uORF) foi mostrado para codificar uma microproteína chamada Mief1-MP. (A) Visão geral do lócus genômico Mief1 . As pontas de flecha nos introns apontam para a direita, indicando que Mief1 é transcrito a partir do fio mais do DNA (foco no PhyloCSF +1, +2 e +3 faixas para determinar o potencial de codificação). O CDS Mief1 principal codifica uma proteína de aminoácidos 463 e é mostrado no painel (B). No entanto, há também um ORF distinto conservado a montante dentro do UTR de 5' de Mief1 que codifica uma microproteína única de 70 aminoácidos chamada Mief1-MP (C). Como visto no Painel C, o Mief1-MP tem seu próprio codon de início e parada conservado dentro do Mief1 5' UTR, e o ORF pontua muito bem na faixa PhyloCSF +1, fornecendo fortes evidências de que ele codifica uma microproteína funcional. Abreviaturas: ORF = quadro de leitura aberto; uORF = ORF upstream; UTR = região não traduzida; CDS = sequência de codificação. Clique aqui para ver uma versão maior desta figura.
| Símbolo | Nível de Conservação de Aminoácidos | Aminoácidos agrupados |
| Asterisco (*) | Resíduo totalmente conservado | Não aplicável (resíduo único e totalmente conservado) |
| Cólon (:) | Grupos com propriedades fortemente semelhantes | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW |
| Período (.) | Grupos com propriedades fracamente semelhantes | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Espaço (sem símbolo) | Sem semelhança | Não aplicável (sem semelhança) |
Tabela 1: Definições de símbolos de consenso para alinhamentos de sequência múltipla gerados por Ômega Clustal. O alinhamento da sequência de várias espécies mostrado na Figura 2 foi gerado usando o Ômega52 Clustal. Abreviaturas: serina (S), threonine (T), alanina (A), asparagina (N), ácido glutamico (E), glutamina (Q), lisina (K), ácido aspartic (D), arginina (R), metionina (R), metionina (E), glutamina (Q), lisina (K), ácido aspartic (D), arginina (R), metionina (R). M), isoleucina (I), leucina (L), fenilalanina (F), histidina (H), tyrosina (Y), triptofano (W), cisteína (C), valina (V), glicina (G), prolina (P).
| Cor da fonte | Propriedade | Resíduo de aminoácido [Abreviação] |
| Vermelho | Pequeno, hidrofóbico | alanina [A], valina [V], fenilalanina [F], proline [P], metorpina [M], isoleucina [I], leucina [L], triptofano [W] |
| Azul | Ácido | ácido aspartic [D], ácido glutamico [E] |
| Magenta | Básico | arginina [R], lysina [K] |
| Verde | Hidroxl, sulfhydryl, amina, +G | serina [S], threonine [T], tyrosine [Y], histidina [H], cisteína [C], asparagine [N], glycina [G], glutamina [Q] |
Tabela 2: Propriedades dos aminoácidos retratados na Figura 2. ClustalOmega 52 foi usado para gerar o alinhamento de sequência múltipla mostrado na Figura 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
O protocolo aqui apresentado fornece instruções detalhadas sobre como interrogar regiões genômicas de interesse para potencial de codificação de microproteínas usando PhyloCSF no Navegador de Genoma UCSCfácil de usar 48,49,50,51. Como detalhado acima, PhyloCSF é um poderoso algoritmo de genômica comparativa que integra modelos filogenéticos e frequências de substituição de codon para identificar assinaturas evolutivas típicas de genes codificadores de proteínas48,49. O PhyloCSF tem sido amplamente utilizado para identificar microproteínas funcionais em regiões genômicas anteriormente anotadas como não codificação 38,39,40,41,42,43,44,45,46,47 , e essa abordagem tem sido demonstrada para superar outros métodos comparativos de genômica para sequências curtas, como microproteínas de até 13 aminoácidos e para pequenos exons de proteínas canônicas 35,48,49. Notavelmente, a utilidade do PhyloCSF como um método robusto para identificar sequências funcionais de codificação de proteínas através da conservação evolutiva se estende além da de espécies vertebradas e invertebradas e foi recentemente aplicada a genomas virais para interrogar com sucesso a capacidade de codificação de proteínas do genoma SARS-CoV-267.
Além de identificar sequências de codificação putativa dentro de RNAs não codificadas anotadas, uma vantagem do PhyloCSF é que ele também pode detectar de forma confiável microproteínas conservadas codificadas por ORFs dentro de regiões não traduzidas anotadas (UTRs) de genes de codificação de proteínas canônicas, incluindo orfs de 5' upstream e 3' downstream (uORFs e dORFs, respectivamente)8,19,66,68 . Por exemplo, a microproteína MIEF1 (MIEF1-MP) está codificada no UTR de 5' do fator de alongamento mitocondrial 1 (MIEF1)66. No caso do MIEF1-MP, observa-se um escore PhyloCSF positivo discrete correspondente ao MIEF1-MP a montante do ORF que codifica MIEF1 (Figura 5). Enquanto algumas microproteínas codificadas uORF interagem diretamente com as proteínas canônicas a jusante em seu mRNA compartilhado, (ex. MIEF1-MP e MIEF1), outras funcionam independentemente da proteína codificada pelo CDSprincipal 66,68. Portanto, ao caracterizar microproteínas codificadas pelo UORF, não se deve supor que elas funcionem através da regulação direta de seu produto proteico a jusante.
Embora o PhyloCSF tenha muitos pontos fortes claros como uma ferramenta para a identificação de sequências conservadas de codificação de microproteínas, é importante reconhecer várias limitações deste método. Em primeiro lugar, enquanto a conservação de sequências sugere fortemente que uma região genômica passou por uma seleção funcional e, portanto, é codificação, a falta de conservação robusta e uma pontuação phyloCSF negativa resultante não descarta definitivamente o potencial de codificação para uma determinada sequência. Em outras palavras, confiar exclusivamente no PhyloCSF pode resultar na supervisão de ORFs traduzidos que não são fortemente conservados, mas ainda produzem microproteínas funcionais. Notavelmente, regiões genômicas com baixos ecores de conservação ou conservação negativa podem corresponder a regiões de codificação específicas de espécies ou aquelas de genes "jovens" evolucionários por divergência sequencial ou nascimento de novos genes 46,69,70,71,72,73,74. Por exemplo, a microproteína o mais rápido possível, que é codificada pelo que se pensava ser o RNA não codificador humano LINC00467, não é pontuada positivamente pelo PhyloCSF porque a sequência de aminoácidos só é conservada em mamíferos superiores75. Além disso, estudos recentes identificaram várias microproteínas específicas do homem, incluindo uma codificada pelo RP3-527G5.1 intergênico, que não gera uma pontuação phyloCSF positiva68,72. A este respeito, a ausência de uma pontuação phyloCSF positiva não pode ser interpretada como prova de uma região não codificadora e deve ser interpretada com cautela.
Uma segunda consideração a ter em mente ao usar o PhyloCSF é que, embora um resultado positivo seja altamente sugestivo da capacidade funcional de seleção e codificação de proteínas, esta linha de evidências não pode ficar sozinha e deve ser validada experimentalmente. Exemplos de métodos que podem ser usados para gerar evidências de apoio para a expressão estável da microproteína incluem a detecção da proteína putativa por espectrometria de massa ou mancha ocidental usando um anticorpo levantado contra a sequência de interesse da microproteína. Alternativamente, uma vez que pode ser desafiador gerar anticorpos confiáveis para microproteínas devido à falta de opções de sequência para a melhor antigenicidade, também é possível usar CRISPR/Cas9 e a via de reparação direcionada à homologia (HDR) para introduzir uma tag epitope no lócus endógeno no quadro com a sequência de microproteína putativa, facilitando assim a detecção da proteína de interesse usando um anticorpo de alta afinidade (e.g., BANDEIRA, HA, V5, Myc)18. Uma limitação final do PhyloCSF para reconhecer é que, embora esteja atualmente integrado em muitos dos conjuntos genômicos comumente usados, incluindo Homo sapiens (hg19 humano, hg38), Mus musculus (mouse mm10, mm39), Gallus gallus (frango, galGal4, galGal6), Drosophila melanogaster (mosca-das-frutas, dm6), Caenorhabditis elegans (nematoides, ce11) e SARS-CoV-2 (wuhCor1), ainda existem muitas espécies que atualmente não podem ser consultadas diretamente no Navegador de Genomas UCSC.
A identificação de domínios conservados ou características de sequência dentro de microproteínas identificadas pode ajudar a aumentar a confiança em sua relevância funcional e fornecer alguma visão sobre sua função putativa. Aqui fornecemos recomendações para ferramentas e recursos específicos que podem ser usados para analisar sequências de aminoácidos de microproteína identificadas em mais detalhes para obter tal percepção. As ferramentas específicas listadas abaixo (e resumidas na Tabela de Materiais) estão disponíveis gratuitamente ao público, e descobrimos que elas são particularmente fáceis de usar e robustas em estudos de microproteína 18,38,39,40,41,47. Além das ferramentas descritas aqui, há uma infinidade de recursos adicionais que podem ser encontrados em portais de recursos bioinformáticas como Expasy (https://www.expasy.org) e EMBL-EBI (https://www.ebi.ac.uk/services/all). No entanto, detalhar as especificidades para cada uma das ferramentas dentro desses repositórios está além do escopo deste artigo. Aqui recomendamos os seguintes recursos.
Primeiro, o TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) analisa sequências proteicas de interesse para a presença de domínios transmembranos. Notavelmente, uma série de microproteínas que foram funcionalmente caracterizadas até agora contêm domínios transmembranos de passagem única, o que facilita sua localização para regiões de membrana e permite sua regulação direta de canais de íons, trocadores e enzimas associadas à membrana30. Em segundo lugar, o National Center for Biotechnology Information (NCBI) Conservado Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) é uma ferramenta popular usada para identificar domínios conservados dentro de sequências de nucleotídeos de proteína ou codificação. Em terceiro lugar, a família proteína (Pfam)78 (http://pfam.xfam.org) fornece alinhamentos e classificações de famílias e domínios proteicos. Em quarto lugar, o WoLF PSORT79 (https://wolfpsort.hgc.jp/) é uma ferramenta que pode ser empregada para prever a localização de proteínas subcelulares. Em quinto lugar, COXPRESdB80 é um banco de dados de co-expressão genética (https://coxpresdb.jp) que fornece relações genéticas co-reguladas para estimar funções genéticas. Finalmente, o SignalP 6.081 é um programa de previsão amplamente utilizado (https://services.healthtech.dtu.dk/service.php?SignalP) que reconhece a presença de uma sequência de peptídeos de sinal e prevê a localização do local do decote.
Em resumo, os métodos descritos aqui podem ser usados para analisar efetivamente regiões genômicas de interesse para o potencial de codificação de proteínas usando PhyloCSF no Navegador de Genoma UCSC. Esses métodos são altamente acessíveis e podem ser facilmente aprendidos e aplicados de forma eficiente por indivíduos sem treinamento prévio ou experiência em bioinformática ou genômica comparativa. Como demonstrado aqui em detalhes, o PhyloCSF é uma ferramenta poderosa que pode ser aplicada como uma análise de primeiro passo para ajudar a distinguir a codificação de proteínas versus genes não codificadores em genomas vertebrados, invertebrados e virais, e os pontos fortes dessa abordagem superam fortemente as fraquezas notadas.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores declaram que não têm interesses financeiros concorrentes.
Acknowledgments
Este trabalho foi apoiado por subsídios dos Institutos Nacionais de Saúde (HL-141630 e HL-160569) e da Cincinnati Children's Research Foundation (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
