

Summary
Le protocole décrit ici fournit des instructions détaillées sur la façon d’analyser les régions génomiques d’intérêt pour le potentiel de codage des microprotéines à l’aide de PhyloCSF sur le navigateur de génome convivial UCSC. En outre, plusieurs outils et ressources sont recommandés pour étudier plus avant les caractéristiques de séquence des microprotéines identifiées afin de mieux comprendre leurs fonctions putatives.
Abstract
Le séquençage de nouvelle génération (NGS) a propulsé le domaine de la génomique vers l’avant et produit des séquences de génome entier pour de nombreuses espèces animales et organismes modèles. Cependant, malgré cette richesse d’informations sur les séquences, les efforts complets d’annotation des gènes se sont avérés difficiles, en particulier pour les petites protéines. Notamment, les méthodes conventionnelles d’annotation des protéines ont été conçues pour exclure intentionnellement les protéines putatives codées par de courts cadres de lecture ouverts (sORF) de moins de 300 nucléotides de longueur afin de filtrer le nombre exponentiellement plus élevé de faux sORF non codants dans tout le génome. En conséquence, des centaines de petites protéines fonctionnelles appelées microprotéines (<100 acides aminés de longueur) ont été classées à tort comme des ARN non codants ou complètement négligées.
Ici, nous fournissons un protocole détaillé pour tirer parti d’outils bioinformatiques gratuits et accessibles au public pour interroger les régions génomiques sur le potentiel de codage des microprotéines basé sur la conservation évolutive. Plus précisément, nous fournissons des instructions étape par étape sur la façon d’examiner la conservation des séquences et le potentiel de codage à l’aide des fréquences de substitution phylogénétique du codon (PhyloCSF) sur le navigateur de génome convivial de l’Université de Californie à Santa Cruz (UCSC). De plus, nous détaillons les étapes pour générer efficacement plusieurs alignements d’espèces de séquences de microprotéines identifiées afin de visualiser la conservation des séquences d’acides aminés et recommandons des ressources pour analyser les caractéristiques des microprotéines, y compris les structures de domaine prédites. Ces outils puissants peuvent être utilisés pour aider à identifier des séquences de codage de microprotéines putatives dans des régions génomiques non canoniques ou pour exclure la présence d’une séquence codante conservée avec un potentiel translationnel dans une transcription d’intérêt non codante.
Introduction
L’identification de l’ensemble complet des éléments codants dans le génome est un objectif majeur depuis le lancement du projet du génome humain et demeure un objectif central pour la compréhension des systèmes biologiques et l’étiologie des maladies génétiques 1,2,3,4. Les progrès des techniques NGS ont conduit à la production de séquences du génome entier pour un grand nombre d’organismes, y compris les vertébrés, les invertébrés, les levures et les plantes5. De plus, les méthodes de séquençage transcriptionnel à haut débit ont révélé la complexité du transcriptome cellulaire et identifié des milliers de nouvelles molécules d’ARN ayant à la fois des fonctions codant pour les protéines et non codantes 6,7. Le décodage de cette grande quantité d’informations de séquence est un processus continu, et des défis subsistent avec des efforts complets d’annotationde gènes 8.
Le développement récent de méthodes de profilage translationnel, y compris le profilage des ribosomes 9,10 et le séquençage des poly-ribosomes11, a fourni des preuves indiquant que des centaines d’événements de traduction non canoniques correspondent à des SORF actuellement non annotés dans tout le génome, avec le potentiel de générer de petites protéines appelées microprotéines ou micropeptides 12,13,14,15,16, 17. Les microprotéines sont apparues comme une nouvelle classe de protéines polyvalentes auparavant négligées par les méthodes standard d’annotation des gènes en raison de leur petite taille (<100 acides aminés) et de l’absence de caractéristiques génétiques classiquescodant pour les protéines 8,12,18,19,20. Les microprotéines ont été décrites dans pratiquement tous les organismes, y compris la levure21,22, les mouches 17,23,24 et les mammifères 25,26,27,28, et il a été démontré qu’elles jouent un rôle essentiel dans divers processus, y compris le développement, le métabolisme et la signalisation du stress 19,20,29, 30,31,32,33,34. Ainsi, il est impératif de continuer à exploiter le génome pour trouver d’autres membres de cette classe de petites protéines fonctionnelles longtemps négligée.
Malgré la reconnaissance généralisée de l’importance biologique des microprotéines, cette classe de gènes reste largement sous-représentée dans les annotations du génome, et leur identification précise continue d’être un défi permanent qui a entravé les progrès dans le domaine. Divers outils de calcul et méthodes expérimentales ont récemment été mis au point pour surmonter les difficultés associées à l’identification des séquences codant pour les microprotéines (discutés en détail dans plusieurs revues complètes 8,35,36,37). De nombreuses études récentes d’identification des microprotéines 38,39,40,41,42,43,44,45,46,47 se sont fortement appuyées sur l’utilisation d’un tel algorithme appelé PhyloCSF 48,49 , une puissante approche génomique comparative qui peut être exploitée pour distinguer les régions du génome codant pour les protéines conservées de celles qui ne sont pas codantes.
Le PhyloCSF compare les fréquences de substitution des codons (LCR) à l’aide d’alignements de nucléotides multi-espèces et de modèles phylogénétiques pour détecter les signatures évolutives de gènes codant pour les protéines. Cette approche empirique basée sur un modèle repose sur la prémisse que les protéines sont principalement conservées au niveau des acides aminés plutôt qu’à la séquence nucléotidique. Par conséquent, les substitutions de codon synonymes, qui codent le même acide aminé, ou les substitutions de codon aux acides aminés ayant des propriétés conservées (c.-à-d. charge, hydrophobicité, polarité) sont notées positivement, tandis que les substitutions non synonymes, y compris les substitutions fausses et absurdes, obtiennent un score négatif. PhyloCSF est formé sur des données du génome entier et s’est avéré efficace pour marquer de courtes portions d’une séquence codante (CDS) isolée de la séquence complète, ce qui est nécessaire lors de l’analyse de microprotéines ou d’exons individuels de gènes codant pour des protéines standard48,49.
Notamment, l’intégration récente des hubs de suivi PhyloCSF dans le Genome Browser 49,50,51 de l’Université de Californie à Santa Cruz (UCSC) permet aux chercheurs de tous horizons d’accéder facilement à une interface conviviale pour interroger les régions génomiques d’intérêt pour le potentiel de codage des protéines. Le protocole décrit ci-dessous fournit des instructions détaillées sur la façon de charger les hubs de suivi PhyloCSF sur le navigateur de génome UCSC et d’interroger ensuite les régions génomiques d’intérêt pour sonder les régions codant pour les protéines à haute confiance (ou l’absence de celles-ci). De plus, dans le cas où un score PhyloCSF positif est observé, des étapes sont délimitées pour analyser davantage le potentiel codant pour les microprotéines et générer efficacement plusieurs alignements d’espèces des séquences d’acides aminés identifiées afin d’illustrer la conservation des séquences inter-espèces. Enfin, plusieurs ressources et outils supplémentaires accessibles au public sont présentés dans la discussion pour étudier les caractéristiques des microprotéines identifiées, y compris les structures de domaine prédites et les connaissances sur la fonction des microprotéines putatives.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Le protocole décrit ci-dessous détaille les étapes à suivre pour charger et naviguer dans les pistes du navigateur PhyloCSF sur le navigateur du génome UCSC (généré par Mudge et al.49). Pour des questions générales concernant le navigateur de génome UCSC, un guide complet de l’utilisateur de Genome Browser peut être trouvé ici: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Chargement du PhyloCSF Track Hub dans le navigateur de génome UCSC
- Ouvrez une fenêtre de navigateur Internet et accédez au navigateur de génome UCSC (https://genome.ucsc.edu/).
- Sous l’en-tête Nos outils , sélectionnez l’option Track Hubs .
REMARQUE: L’option Track Hubs se trouve également sous l’onglet Mes données . - Dans l’onglet Hubs publics , tapez PhyloCSF dans la zone Termes de recherche. Cliquez sur le bouton Rechercher dans les hubs publics .
- Connectez-vous à PhyloCSF en cliquant sur le bouton Connecter pour le nom du hub PhyloCSF (Description: Potentiel évolutif codant pour les protéines tel que mesuré par PhyloCSF).
REMARQUE: Ce moyeu de piste se chargera sur de nombreux assemblages, y compris l’homme (hg19 et hg38) et la souris (mm10 et mm39). - Après avoir cliqué sur se connecter, attendez d’être redirigé vers la page UCSC Genome Browser Gateway (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Naviguer vers les gènes d’intérêt à l’aide d’identificateurs de gènes
- Sélectionnez l’espèce et l’assemblage du génome à interroger. Pour interroger une autre espèce (par exemple, la souris), sélectionnez l’espèce d’intérêt sous l’en-tête Parcourir/Sélectionner une espèce en cliquant sur l’icône appropriée, ou tapez l’espèce dans la zone de texte qui dit: Entrez l’espèce, le nom commun ou l’ID d’assemblage.
REMARQUE : L’assemblage est répertorié directement sous l’en-tête Rechercher une position . En règle générale, la valeur par défaut est l’assemblage humain (par exemple, décembre 2009 [GRCh37/hg19]). - Choisissez l’assemblage à rechercher sous l’en-tête Rechercher une position à l’aide du menu déroulant.
- Entrez la position, le symbole du gène ou les termes de recherche dans la zone Position/Terme de recherche et cliquez sur Aller pour accéder à un gène d’intérêt dans le navigateur de génomes.
- Si la recherche a abouti à plusieurs correspondances, attendez d’être redirigé vers une page qui nécessite la sélection d’une position d’intérêt. Cliquez sur le gène approprié qui vous intéresse.
3. Naviguer vers les régions génomiques d’intérêt à l’aide d’informations de séquence
- Accédez au navigateur de génomes UCSC (https://genome.ucsc.edu/) et sélectionnez l’outil d’alignement de type BLAST (BLAT) sous l’en-tête Nos outils pour interroger une séquence d’ADN ou de protéine spécifique. Vous pouvez également placer le curseur sur l’onglet Outils et sélectionner l’option Blat ou suivre ce lien : https://genome.ucsc.edu/cgi-bin/hgBlat.
- Sélectionnez l’espèce (génome) et l’assemblage qui vous intéressent à l’aide des menus déroulants.
- Définissez le type de requête à l’aide du menu déroulant.
- Collez la séquence d’intérêt dans la zone de texte BLAT Search Genome et cliquez sur Envoyer.
- Cliquez sur le lien du navigateur sous la rubrique ACTIONS pour accéder à la région génomique qui vous intéresse.
4. Identification des SORF conservés à l’aide des données de suivi PhyloCSF
- Scannez visuellement la zone d’intérêt génomique pour obtenir une note positive des régions PhyloCSF (Figure 1).
REMARQUE: Pour une explication détaillée de la façon d’interpréter visuellement les scores PhyloCSF sur le navigateur de génome UCSC, consultez la section des résultats représentatifs ci-dessous. - Utilisez la fonction de zoom pour agrandir les régions d’intérêt afin d’examiner les caractéristiques de séquence et de rechercher des codons de démarrage/arrêt. Pour effectuer un zoom avant manuellement, maintenez la touche Maj enfoncée et maintenez enfoncé le bouton de la souris tout en faisant glisser la région d’intérêt. Vous pouvez également utiliser les boutons de zoom avant et de zoom arrière en haut de la page pour naviguer (des options de zoom 1,5x, 3x, 10x ou de base sont disponibles).
REMARQUE: Avant d’utiliser les boutons de zoom avant / zoom arrière , il est nécessaire de repositionner le gène de sorte que la région d’intérêt soit au milieu de l’écran. Pour effectuer cette action, cliquez sur l’image et faites-la glisser vers la gauche ou la droite pour déplacer la région génomique horizontalement comme vous le souhaitez ou utilisez les flèches de déplacement en haut de la page. - Effectuez un zoom avant jusqu’à ce que la séquence nucléotidique (base) soit visible.
REMARQUE: La séquence nucléotidique apparaîtra directement au-dessus du score PhyloCSF lissé +1. - Scannez visuellement la séquence nucléotidique près du début et de la fin des régions PhyloCSF à score positif pour identifier les codons putatifs start (ATG) et stop (TGA/TAA/TAG).
REMARQUE: Si le gène d’intérêt est sur le brin moins de l’ADN, les codons de départ et d’arrêt seront le complément inverse (c.-à-d. CAT pour le codon de départ et TCA / TTA / CTA pour le codon stop).
5. Affichage des régions homologues dans d’autres génomes
- Passez la souris sur l’en-tête Affichage en haut de la page et cliquez sur l’option Dans d’autres génomes (Convertir ).
- Définissez le génome d’intérêt à l’aide du menu déroulant sous l’en-tête Nouveau génome .
- Sélectionnez l’assemblage génomique qui vous intéresse à l’aide du menu déroulant sous l’en-tête Nouvel assemblage , puis cliquez sur le bouton Envoyer .
- Une fois que le navigateur renvoie une liste de régions du nouvel assemblage présentant une similitude, cliquez sur le lien de position du chromosome pour accéder à la région homologue d’intérêt.
REMARQUE : Le pourcentage de bases totales (nucléotides) et la portée couverte par la région seront définis pour chaque région énumérée. Plus le pourcentage de bases correspondantes est élevé, plus la conservation est élevée pour la région d’intérêt. - Suivez les mêmes stratégies de navigation détaillées à la section 4 pour analyser la séquence.
6. Génération d’alignements de séquences multi-espèces pour les microprotéines d’intérêt
- Cliquez sur le gène d’intérêt dans la piste GENCODE sur le navigateur de génome UCSC (indiqué à la figure 1A avec une boîte bleue) pour accéder à la page de description du gène.
- Sous l’en-tête Séquence et liens vers outils et bases de données , cliquez sur le lien dans le tableau qui lit Autres espèces FASTA.
- Cliquez sur les cases associées aux espèces d’intérêt pour les sélectionner. Cliquez sur Soumettre. Copiez et collez les séquences apparaissant en bas de page au format FASTA dans un document de traitement de texte.
- Ouvrez une deuxième fenêtre de navigateur et accédez à l’outil Clustal Omega Multiple Sequence Alignment tool 52 sur le site Web de l’Institut européen de bioinformatique (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Collez les fichiers de séquence qui se trouvent encore dans le Presse-papiers dans la zone de l’ÉTAPE 1 qui lit les séquences dans n’importe quel format pris en charge. Faites défiler jusqu’au bas de la page et cliquez sur Soumettre. Regardez ci-dessous les résultats alignés (en police noire) pour les symboles qui indiquent le degré de conservation de chaque acide aminé (les symboles sont définis dans le tableau 1).
REMARQUE : la génération de l’alignement peut prendre plusieurs minutes. - Pour afficher les propriétés des acides aminés en couleur, cliquez sur le lien Afficher les couleurs directement au-dessus des séquences pour colorer les acides aminés en fonction de leurs propriétés (définies dans le tableau 2).
- Copiez et collez l’alignement de séquence dans un programme de traitement de texte ou de diaporama pour générer une figure ou un fichier d’illustration (par exemple, figure 2).
REMARQUE: Utilisez une police mono-espacée pour l’alignement, telle que Courier. - Pour afficher d’autres résultats de la page de résultats de Clustal Omega , cliquez sur les onglets appropriés (c.-à-d. Arbre guide ou Arbre phylogénétique).
- Cliquez sur l’onglet Visionneuses de résultats pour obtenir des options permettant d’afficher les informations de séquence à l’aide de Jalview, un programme gratuit spécialisé dans l’édition, la visualisation et l’analyse d’alignement de séquencesmultiples 55, ou pour accéder à des liens directs vers MView et Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Ici, nous utiliserons la microprotéine mitoréguline validée (Mtln) comme exemple pour démontrer comment un sORF conservé générera un score PhyloCSF positif qui peut être facilement visualisé et analysé sur le navigateur de génome UCSC. La mitoréguline était auparavant annotée en tant qu’ARN non codant (anciennement ID du gène humain LINC00116 et ID du gène de la souris 1500011K16Rik). La génomique comparative et les méthodes d’analyse de conservation des séquences ont joué un rôle essentiel dans sa découverte initiale 40,57,58,59,60,61, soulignant la force de ces méthodes. Pour cet exemple, l’assemblage de la souris GRCm38/mm10 (décembre 2011) sera utilisé. La recherche peut être effectuée à l’aide des identificateurs de gènes (mitoregulin, Mtln) ou de la position du gène (chr2:127,791,364-127,792,496) comme décrit dans la section 2 du protocole. Alternativement, la séquence d’acides aminés pour la mitoréguline (illustrée à la figure 2) peut être recherchée à l’aide de l’outil BLAT (décrit dans la section 3 du protocole).
Un écran similaire à celui illustré à la figure 1A apparaîtra avec le PhyloCSF Track Hub visible en haut de l’écran. Les pistes PhyloCSF lissées (lissées avec un modèle de Markov caché définissant une probabilité que chaque codon code) sont représentées comme six pistes totales, avec trois pistes correspondant au brin plus de l’ADN (représenté en vert comme PhyloCSF +1, +2 et +3) et trois pistes correspondant au brin moins de l’ADN (représenté en rouge comme PhyloCSF -1, -2 et -3). Ces pistes représentent les trois cadres de lecture potentiels pour le gène d’intérêt dans chaque direction. Dans la fenêtre du navigateur, les exons sont représentés sous forme de rectangles bleus reliés par de fines lignes horizontales bleues, qui représentent les introns. Les pointes de flèche sur les régions introniques indiquent dans quelle direction le gène est transcrit (et donc, sur quel brin se concentrer pour le score PhyloCSF). Pour l’exemple de Mtln dans la figure 1, les pointes de flèche introniques pointent vers la gauche. Par conséquent, le gène Mtln est transcrit à partir du brin moins de l’ADN, et le score PhyloCSF pertinent est représenté dans les pistes -1, -2 et -3 (en rouge).
Chaque piste PhyloCSF est représentée sous la forme d’une fine ligne noire avec des régions de notation négatives représentées en vert clair / rouge sous la ligne et des régions de notation positives indiquées en vert foncé / rouge au-dessus de la ligne. Comme décrit dans l’introduction, un score PhyloCSF positif indique une région conservée qui est susceptible de coder. Notez que pour les régions codant pour les protéines avec une conservation de séquence particulièrement élevée, elles obtiennent souvent également un score positif sur le brin antisens; cependant, le score PhyloCSF est généralement plus élevé sur le bon brin. Par exemple, cela peut être vu dans la figure 1 pour Mtln où la séquence de codage correcte obtient des scores très élevés dans la piste PhyloCSF -1, et le brin antisens (piste PhyloCSF +2) génère également un score positif. Comme le montre la figure 1A (indiquée par une boîte noire), il y a une région dans le premier exon de Mtln qui obtient un score très élevé sur la piste PhyloCSF -1, ce qui suggère que cela peut correspondre à une région codante. Pour examiner cette région plus en détail, il est utile de zoomer et d’agrandir la région (Figure 1B). Comme le montre la figure 1C, D, la région de notation positive dans le premier exon de Mtln commence directement sur un codon de départ (figure 1C) et se termine à un codon d’arrêt (figure 1D), ce qui indique que cet ORF est hautement conservé et suggère fortement qu’il s’agit d’un ORF codant. Comme Mtln est sur le brin moins de l’ADN, les codons de départ et d’arrêt sont représentés comme le complément inverse du codon (c’est-à-dire que le codon de départ ATG est représenté comme CAT [Figure 1C] et le codon stop TGA est représenté comme TCA [Figure 1D]).
En plus d’utiliser PhyloCSF pour rechercher des régions conservées avec un potentiel de codage de microprotéines, cette technique peut également être appliquée comme une analyse de premier passage d’ARN putatifs non codants pour exclure la présence d’un ORF conservé, fournissant ainsi un support pour une annotation non codante. Par exemple, l’analyse de l’ARNnc bien caractérisé HOTAIR62,63 à l’aide de PhyloCSF montre un score négatif sur l’ensemble du gène sur les six pistes (Figure 3), indiquant fortement un manque de conservation de la séquence et confirmant que HOTAIR est correctement annoté en tant qu’ARN non codant.
Comme le montre clairement la figure 1, l’ensemble de l’ORF codant pour la mitoreguline est situé dans un seul exon, produisant ainsi une lecture simple et directe par PhyloCSF avec une seule région de notation positive ininterrompue. Cependant, les données du hub de suivi PhyloCSF ne sont pas toujours aussi claires et faciles à interpréter. Par exemple, la microprotéine mitolamban/Stmp1/Mm47 codée par la souris 1810058I24Rik gène 47,64,65 représente un ORF conservé qui s’étend sur trois exons (Figure 4A), et le score positif PhyloCSF passe de la piste +2 dans l’exon 1 (Figure 4B) à la piste +3 dans l’exon 2 (Figure 4C), puis revient à la piste +2 dans l’exon 3 (Figure 4D). ). Bien qu’à première vue, cela semble déroutant, l’explication est assez simple. PhyloCSF note les six cadres de lecture potentiels (trois sur le brin plus de l’ADN et trois sur le brin moins) des régions génomiques sans tenir compte de l’architecture exon/intron spécifique pour chaque gène. Par conséquent, il conserve les informations de séquence intronique dans la périodicité des 3 nucléotides des cadres de lecture. Ainsi, si un intron contient un nombre de nucléotides qui n’est pas divisible par trois (c.-à-d. trois nucléotides/codon), le cadre de lecture PhyloCSF sautera d’une piste à l’autre.
Enfin, PhyloCSF peut également être utilisé efficacement pour identifier plusieurs ORF codants distincts au sein d’une seule molécule d’ARN. Par exemple, la microprotéine MIEF1 (MIEF1-MP) est codée dans l’UTR 5' du facteur d’allongement mitochondrial 1 (MIEF1)66 (Figure 5). Lorsque la région génomique MIEF1 est analysée par PhyloCSF, un score PhyloCSF positif discret correspondant au MIEF1-MP (Figure 5C) peut être facilement observé en amont du CDS principal pour MIEF1 (Figure 5B). Une discussion plus approfondie sur MIEF1 et ses microprotéines associées (MIEF1-MP) est fournie ci-dessous dans la discussion avec un résumé des forces et des faiblesses des méthodes et des protocoles décrits dans cet article.

Figure 1 : L’analyse PhyloCSF du gène de la mitoreguline (Mtln) indique une région de conservation de séquence élevée correspondant à une microprotéine validée. (A) Des captures d’écran du navigateur de génome UCSC et des pistes PhyloCSF montrent que Mtln contient deux exons et un seul intron. Les pointes de flèche dans l’intron pointent vers la gauche, indiquant que le gène Mtln est transcrit à partir du brin moins de l’ADN, et les scores PhyloCSF pertinents sont donc indiqués dans les pistes -1, -2 et -3 (en rouge). La séquence complète de codage de la mitoréguline est contenue dans l’exon 1 et obtient un score élevé sur la piste PhyloCSF -1 (B). Un codon de départ conservé peut être clairement observé au début de la région de notation positive dans la piste PhyloCSF -1 (C), qui est mise en évidence par une boîte verte (CAT, atG à complément inversé). De plus, un codon d’arrêt conservé (TCA, TGA à complément inversé) est indiqué par une boîte rouge dans le panneau (D), qui s’aligne sur la fin de la région PhyloCSF à score positif. Des informations détaillées sur le gène Mtln peuvent être trouvées en cliquant sur l’identifiant du gène Mtln dans la boîte bleue (illustrée dans le panneau A). Il est à noter que les régions codant pour des protéines hautement conservées obtiennent souvent des scores positifs sur le brin antisens (vu ici dans la piste PhyloCSF +2 pour Mtln). Cependant, le score PhyloCSF est généralement plus élevé sur le brin correct (la piste PhyloCSF -1 dans cet exemple). Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 2 : Alignement de séquences multi-espèces de la microprotéine mitoréguline générée à l’aide du programme Clustal Omega. Les séquences d’acides aminés de mitoreguline pour les huit espèces indiquées ont été extraites comme détaillé dans la section 6 du protocole et alignées avec l’outil d’alignement de séquences multiples Clustal Omega. Les propriétés des acides aminés sont indiquées par la couleur (rouge, petit/hydrophobe; bleu, acide; magenta, basique; vert, hydroxl/sulfhydryle/amine) (définie plus en détail dans le tableau 2). Les symboles sous les acides aminés indiquent le degré de conservation (astérisques, résidus entièrement conservés; côlons, acides aminés aux propriétés fortement similaires; périodes, conservation entre groupes de propriétés faiblement similaires) (détaillé en détail dans le tableau 1). Veuillez cliquer ici pour voir une version agrandie de cette figure.
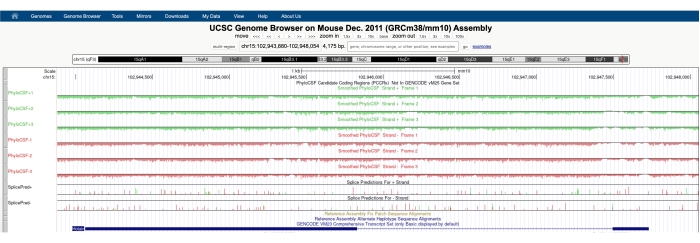
Figure 3 : Une capture d’écran des traces PhyloCSF pour l’ARN long non codant validé Hotair montre un manque de conservation de la séquence dans l’ensemble de son locus génomique. Les pointes de flèches dans la région intronique de Hotair pointent vers la gauche, indiquant que l’ARNnc est transcrit à partir du brin négatif de l’ADN, et donc les pistes PhyloCSF -1, -2 et -3 devraient être au centre de l’analyse. Notez que le score PhyloCSF est négatif dans l’ensemble du gène (pour les six pistes), ce qui indique un manque de conservation de la séquence, ce qui soutient son annotation appropriée en tant qu’ARN non codant. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 4 : Analyse PhyloCSF du gène 1810058I24Rik de la souris, qui code pour la microprotéine mitolamban/Stmp1/Mm47. (A) Le gène 1810058I24Rik de la souris est composé de trois exons, et les pointes de flèche dans les régions introniques pointent vers la droite, indiquant qu’il est transcrit sur le brin plus de l’ADN et donc les pistes PhyloCSF +1, +2 et +3 doivent être analysées. La séquence de codage des microprotéines conservées couvre les trois exons, commençant par l’exon 1 (B), lisant à travers l’exon 2 (C) et se terminant par l’exon 3 (D). Notez que le score positif de PhyloCSF se trouve sur la piste +2 dans l’exon 1, la piste +3 dans l’exon 2 et la piste +2 dans l’exon 1. La raison du déplacement du score positif d’une piste à l’autre est que PhyloCSF analyse les six cadres de lecture potentiels de la séquence d’ADN indépendamment de la structure exon/intron du gène. Par conséquent, un intron contenant un nombre de nucléotides qui n’est pas divisible par trois (trois nucléotides/codon) entraînera un déplacement du cadre de lecture vers une piste différente. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 5 : L’analyse du locus génomique Mief1 avec PhyloCSF identifie une région avec un potentiel codant pour les protéines dans l’UTR 5' qui est indépendante du CDS Mief1 principal sur l’ARN partagé. Il a été démontré que ce BLR en amont conservé (uORF) code une microprotéine nommée Mief1-MP. (A) Vue d’ensemble du locus génomique Mief1. Les pointes de flèche dans les introns pointent vers la droite, indiquant que Mief1 est transcrit à partir du brin plus d’ADN (concentrez-vous sur les pistes PhyloCSF +1, +2 et +3 pour déterminer le potentiel de codage). Le CDS mief1 principal code une protéine d’acide aminé 463 et est montré dans le panneau (B). Cependant, il existe également un ORF distinct conservé en amont dans l’UTR de 5' de Mief1 qui code une microprotéine unique de 70 acides aminés appelée Mief1-MP (C). Comme on l’a vu dans le panneau C, le Mief1-MP a son propre codon de démarrage et d’arrêt conservé dans le Mief1 5' UTR, et l’ORF obtient de très bons résultats sur la piste PhyloCSF +1, fournissant des preuves solides qu’il code une microprotéine fonctionnelle. Abréviations : ORF = cadre de lecture ouvert; uORF = ORF en amont; UTR = région non traduite; CDS = séquence de codage. Veuillez cliquer ici pour voir une version agrandie de cette figure.
| Symbole | Niveau de conservation des acides aminés | Acides aminés groupés |
| Astérisque (*) | Résidus entièrement conservés | Sans objet (résidu unique entièrement conservé) |
| Côlon (:) | Groupes ayant des propriétés fortement similaires | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW |
| Période (.) | Groupes ayant des propriétés faiblement similaires | L’ASC; VTT; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Espace (pas de symbole) | Aucune similitude | Sans objet (pas de similitude) |
Tableau 1 : Définitions des symboles de consensus pour les alignements de séquences multiples générés par Clustal Omega. L’alignement des séquences multi-espèces illustré à la figure 2 a été généré à l’aide de Clustal Omega52. Abréviations : sérine (S), thréonine (T), alanine (A), asparagine (N), acide glutamique (E), glutamine (Q), lysine (K), acide aspartique (D), arginine (R), méthionine (M), isoleucine (I), leucine (L), phénylalanine (F), histidine (H), tyrosine (Y), tryptophane (W), cystéine (C), valine (V), glycine (G), proline (P).
| Couleur de police | Propriété | Résidu d’acides aminés [Abréviation] |
| Rouge | Petit, hydrophobe | alanine [A], valine [V], phénylalanine [F], proline [P], méthionine [M], isoleucine [I], leucine [L], tryptophane [W] |
| Bleu | Acide | acide aspartique [D], acide glutamique [E] |
| Magenta | Basique | arginine [R], lysine [K] |
| Vert | Hydroxl, sulfhydryle, amine, +G | sérine [S], thréonine [T], tyrosine [Y], histidine [H], cystéine [C], asparagine [N], glycine [G], glutamine [Q] |
Tableau 2 : Propriétés des acides aminés représentés à la figure 2. Clustal Omega52 a été utilisé pour générer l’alignement de séquences multiples illustré à la figure 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Le protocole présenté ici fournit des instructions détaillées sur la façon d’interroger les régions génomiques d’intérêt pour le potentiel de codage des microprotéines à l’aide de PhyloCSF sur le navigateur de génome convivial UCSC 48,49,50,51. Comme détaillé ci-dessus, PhyloCSF est un puissant algorithme de génomique comparative qui intègre des modèles phylogénétiques et des fréquences de substitution de codon pour identifier les signatures évolutives typiques des gènes codant pour les protéines48,49. PhyloCSF a été largement utilisé pour identifier les microprotéines fonctionnelles dans les régions génomiques précédemment annotées comme non codantes 38,39,40,41,42,43,44,45,46,47 , et il a été démontré que cette approche surpasse d’autres méthodes de génomique comparative pour des séquences courtes telles que des microprotéines aussi petites que 13 acides aminés et pour de petits exons de protéines canoniques 35,48,49. Notamment, l’utilité du PhyloCSF en tant que méthode robuste pour identifier les séquences fonctionnelles codant pour les protéines via la conservation évolutive s’étend au-delà de celle des espèces de vertébrés et d’invertébrés et a même été récemment appliquée aux génomes viraux pour interroger avec succès la capacité de codage des protéines du génome sars-CoV-267.
En plus d’identifier des séquences codantes putatives dans des ARN annotés non codants, un avantage de PhyloCSF est qu’il peut également détecter de manière fiable les microprotéines conservées codées par des ORF dans des régions annotées non traduites (UTR) de gènes codant pour des protéines canoniques, y compris les ORF 5' en amont et 3' en aval (uORF et dORF, respectivement)8,19,66,68 . Par exemple, la microprotéine MIEF1 (MIEF1-MP) est codée dans l’UTR 5' du facteur d’allongement mitochondrial 1 (MIEF1)66. Dans le cas du MIEF1-MP, un score PhyloCSF positif discret correspondant au MIEF1-MP est observé en amont de l’ORF qui code MIEF1 (Figure 5). Alors que certaines microprotéines codées uORF interagissent directement avec les protéines canoniques en aval sur leur ARNm partagé (ex. MIEF1-MP et MIEF1), d’autres fonctionnent indépendamment de la protéine codée par le CDSprincipal 66,68. Par conséquent, lors de la caractérisation des microprotéines codées uORF, il ne faut pas supposer qu’elles fonctionnent via la régulation directe de leur produit protéique en aval.
Bien que PhyloCSF ait de nombreux atouts clairs en tant qu’outil d’identification des séquences codant pour les microprotéines conservées, il est important de reconnaître plusieurs limites de cette méthode. Tout d’abord, alors que la conservation des séquences suggère fortement qu’une région génomique a subi une sélection fonctionnelle et code donc, un manque de conservation robuste et un score PhyloCSF négatif qui en résulte n’excluent pas définitivement le potentiel de codage pour une séquence donnée. En d’autres termes, s’appuyer exclusivement sur PhyloCSF peut entraîner la surveillance des ORF traduits qui ne sont pas fortement conservés mais qui produisent tout de même des microprotéines fonctionnelles. Notamment, les régions génomiques avec des scores de conservation faibles ou négatifs pourraient correspondre à des régions codantes spécifiques à l’espèce ou à celles de gènes « jeunes » évolutifs via la divergence de séquence ou la naissance de gènes de novo 46,69,70,71,72,73,74. Par exemple, la microprotéine ASAP, qui est codée par ce que l’on pensait autrefois être l’ARN humain non codant LINC00467, n’est pas notée positivement par PhyloCSF parce que la séquence d’acides aminés n’est conservée que chez les mammifères supérieurs75. De plus, des études récentes ont identifié plusieurs microprotéines spécifiques à l’homme, dont une codée par l’ARNnc intergénique RP3-527G5.1, qui ne génère pas un score PhyloCSF positifde 68,72. À cet égard, l’absence d’un score PhyloCSF positif ne peut être interprétée comme la preuve d’une région non codante et doit être interprétée avec prudence.
Une deuxième considération à garder à l’esprit lors de l’utilisation de PhyloCSF est que même si un score positif est fortement évocateur de la sélection fonctionnelle et de la capacité de codage des protéines, cette ligne de preuve ne peut pas être isolée et doit être validée expérimentalement. Des exemples de méthodes qui peuvent être utilisées pour générer des preuves à l’appui de l’expression stable des microprotéines comprennent la détection de la protéine putative par spectrométrie de masse ou western blotting à l’aide d’un anticorps soulevé contre la séquence de microprotéines d’intérêt. Alternativement, comme il peut être difficile de générer des anticorps fiables pour les microprotéines en raison du manque de choix de séquence pour une antigénicité optimale, il est également possible d’utiliser CRISPR / Cas9 et la voie de réparation dirigée par homologie (HDR) pour introduire une étiquette épitope dans le locus endogène dans le cadre de la séquence de microprotéines putative, facilitant ainsi la détection de la protéine d’intérêt à l’aide d’un anticorps à haute affinité (par exemple, FLAG, HA, V5, Myc)18. Une dernière limite de PhyloCSF à reconnaître est que, bien qu’il soit actuellement intégré dans de nombreux assemblages génomiques couramment utilisés, y compris Homo sapiens (hg19 humain, hg38), Mus musculus (souris mm10, mm39), Gallus gallus (poulet, galGal4, galGal6), Drosophila melanogaster (mouche des fruits, dm6), Caenorhabditis elegans (nématodes, ce11) et sars-CoV-2 (wuhCor1), il existe encore de nombreuses espèces qui ne peuvent actuellement pas être interrogées directement sur le navigateur du génome de l’UCSC.
L’identification de domaines conservés ou de caractéristiques de séquence dans les microprotéines identifiées peut aider à accroître la confiance dans leur pertinence fonctionnelle et à fournir un aperçu de leur fonction putative. Ici, nous fournissons des recommandations pour des outils et des ressources spécifiques qui peuvent être utilisés pour analyser plus en détail les séquences d’acides aminés de microprotéines identifiées afin d’obtenir de telles informations. Les outils spécifiques énumérés ci-dessous (et résumés dans le tableau des matériaux) sont mis gratuitement à la disposition du public, et nous les avons trouvés particulièrement conviviaux et robustes dans les études sur les microprotéines 18,38,39,40,41,47. Au-delà des outils décrits ici, il existe une multitude de ressources supplémentaires que l’on peut trouver dans les portails de ressources bioinformatiques tels que Expasy (https://www.expasy.org) et EMBL-EBI (https://www.ebi.ac.uk/services/all). Toutefois, détailler les spécificités de chacun des outils de ces référentiels dépasse le cadre de cet article. Ici, nous recommandons les ressources suivantes.
Tout d’abord, TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) analyse les séquences protéiques d’intérêt pour la présence de domaines transmembranaires. Notamment, un certain nombre de microprotéines qui ont été fonctionnellement caractérisées jusqu’à présent contiennent des domaines transmembranaires à passage unique, ce qui facilite leur localisation dans les régions membranaires et permet leur régulation directe des canaux ioniques, des échangeurs et des enzymes associées à la membrane30. Deuxièmement, le National Center for Biotechnology Information (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) est un outil populaire utilisé pour identifier les domaines conservés dans les séquences de protéines ou de nucléotides codants. Troisièmement, la base de données Protein Family (Pfam)78 (http://pfam.xfam.org) fournit des alignements et des classifications des familles et des domaines de protéines. Quatrièmement, WoLF PSORT79 (https://wolfpsort.hgc.jp/) est un outil qui peut être utilisé pour prédire la localisation des protéines subcellulaires. Cinquièmement, COXPRESdB80 est une base de données de co-expression génique (https://coxpresdb.jp) qui fournit des relations génétiques co-régulées pour estimer les fonctions des gènes. Enfin, SignalP 6.081 est un programme de prédiction largement utilisé (https://services.healthtech.dtu.dk/service.php?SignalP) qui reconnaît la présence d’une séquence peptidique signal et prédit l’emplacement du site de clivage.
En résumé, les méthodes décrites ici peuvent être utilisées pour analyser efficacement les régions génomiques d’intérêt pour le potentiel de codage des protéines en utilisant PhyloCSF sur le navigateur de génome UCSC. Ces méthodes sont très accessibles et peuvent être facilement apprises et appliquées efficacement par des personnes sans formation ou expertise préalables en bioinformatique ou en génomique comparative. Comme démontré ici en détail, PhyloCSF est un outil puissant qui peut être appliqué comme une analyse de premier passage pour aider à distinguer les gènes codant pour les protéines des gènes non codants dans les génomes des vertébrés, des invertébrés et des viraux, et les forces de cette approche l’emportent largement sur les faiblesses notées.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs déclarent qu’ils n’ont pas d’intérêts financiers concurrents.
Acknowledgments
Ce travail a été soutenu par des subventions des National Institutes of Health (HL-141630 et HL-160569) et de la Cincinnati Children’s Research Foundation (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
