

Summary
El protocolo descrito aquí proporciona instrucciones detalladas sobre cómo analizar las regiones genómicas de interés para el potencial de codificación de microproteínas utilizando PhyloCSF en el navegador del genoma UCSC fácil de usar. Además, se recomiendan varias herramientas y recursos para investigar más a fondo las características de secuencia de las microproteínas identificadas para obtener información sobre sus supuestas funciones.
Abstract
La secuenciación de próxima generación (NGS) ha impulsado el campo de la genómica hacia adelante y ha producido secuencias del genoma completo para numerosas especies animales y organismos modelo. Sin embargo, a pesar de esta gran cantidad de información de secuencia, los esfuerzos integrales de anotación de genes han demostrado ser un desafío, especialmente para las proteínas pequeñas. En particular, los métodos convencionales de anotación de proteínas se diseñaron para excluir intencionalmente las proteínas putativas codificadas por marcos de lectura abiertos cortos (sORF) de menos de 300 nucleótidos de longitud para filtrar el número exponencialmente mayor de sORFs espurios no codificantes en todo el genoma. Como resultado, cientos de pequeñas proteínas funcionales llamadas microproteínas (<100 aminoácidos de longitud) se han clasificado incorrectamente como ARN no codificantes o se han pasado por alto por completo.
Aquí proporcionamos un protocolo detallado para aprovechar las herramientas bioinformáticas gratuitas y disponibles públicamente para consultar las regiones genómicas en busca de potencial de codificación de microproteínas basado en la conservación evolutiva. Específicamente, proporcionamos instrucciones paso a paso sobre cómo examinar la conservación de la secuencia y el potencial de codificación utilizando frecuencias de sustitución de codones filogenéticos (PhyloCSF) en el navegador del genoma fácil de usar de la Universidad de California en Santa Cruz (UCSC). Además, detallamos los pasos para generar de manera eficiente alineaciones de múltiples especies de secuencias de microproteínas identificadas para visualizar la conservación de la secuencia de aminoácidos y recomendamos recursos para analizar las características de las microproteínas, incluidas las estructuras de dominio predichas. Estas poderosas herramientas se pueden utilizar para ayudar a identificar secuencias supuestas de codificación de microproteínas en regiones genómicas no canónicas o para descartar la presencia de una secuencia de codificación conservada con potencial de traducción en una transcripción no codificante de interés.
Introduction
La identificación del conjunto completo de elementos codificantes en el genoma ha sido un objetivo importante desde el inicio del Proyecto Genoma Humano, y sigue siendo un objetivo central hacia la comprensión de los sistemas biológicos y la etiología de las enfermedades de base genética 1,2,3,4. Los avances en las técnicas ngs han llevado a la producción de secuencias del genoma completo para un gran número de organismos, incluidos vertebrados, invertebrados, levaduras y plantas5. Además, los métodos de secuenciación transcripcional de alto rendimiento han revelado aún más la complejidad del transcriptoma celular e identificado miles de nuevas moléculas de ARN con funciones codificantes y no codificantesde proteínas 6,7. La decodificación de esta gran cantidad de información de secuencia es un proceso continuo, y siguen existiendo desafíos con los esfuerzos integrales de anotaciónde genes 8.
El reciente desarrollo de métodos de perfiles traslacionales, incluidos el perfil de ribosomas 9,10 y la secuenciación de poli ribosomas11, han proporcionado evidencia que indica que cientos de eventos de traducción no canónica se asignan a sORF actualmente no anotados en todo el genoma, con el potencial de generar pequeñas proteínas llamadas microproteínas o micropéptidos 12,13,14,15,16, 17. Las microproteínas han surgido como una nueva clase de proteínas versátiles previamente pasadas por alto por los métodos estándar de anotación de genes debido a su pequeño tamaño (<100 aminoácidos) y la falta de características clásicas de genes codificantes de proteínas 8,12,18,19,20. Las microproteínas se han descrito en prácticamente todos los organismos, incluyendo la levadura21,22, las moscas 17,23,24 y los mamíferos 25,26,27,28, y se ha demostrado que desempeñan un papel crítico en diversos procesos, incluyendo el desarrollo, el metabolismo y la señalización de estrés 19,20,29, 30,31,32,33,34. Por lo tanto, es imperativo continuar extrayendo el genoma para miembros adicionales de esta clase de proteínas pequeñas funcionales que se han pasado por alto durante mucho tiempo.
A pesar del reconocimiento generalizado de la importancia biológica de las microproteínas, esta clase de genes sigue estando muy poco representada en las anotaciones del genoma, y su identificación precisa sigue siendo un desafío continuo que ha obstaculizado el progreso en el campo. Recientemente se han desarrollado varias herramientas computacionales y métodos experimentales para superar las dificultades asociadas con la identificación de secuencias codificantes de microproteínas (discutidas ampliamente en varias revisiones exhaustivas 8,35,36,37). Muchos estudios recientes de identificación de microproteínas 38,39,40,41,42,43,44,45,46,47 se han basado en gran medida en el uso de uno de estos algoritmos llamado PhyloCSF 48,49 , un poderoso enfoque de genómica comparativa que se puede aprovechar para distinguir las regiones codificantes de proteínas conservadas del genoma de las que no codifican.
PhyloCSF compara las frecuencias de sustitución de codones (CSF) utilizando alineaciones de nucleótidos de múltiples especies y modelos filogenéticos para detectar firmas evolutivas de genes codificantes de proteínas. Este enfoque empírico basado en modelos se basa en la premisa de que las proteínas se conservan principalmente a nivel de aminoácidos en lugar de la secuencia de nucleótidos. Por lo tanto, las sustituciones de codones sinónimos, que codifican el mismo aminoácido, o las sustituciones de codones a aminoácidos con propiedades conservadas (es decir, carga, hidrofobicidad, polaridad) se puntúan positivamente, mientras que las sustituciones no sinónimas, incluidas las sustituciones sin sentido y sin sentido, puntúan negativamente. PhyloCSF está entrenado en datos de genoma completo y ha demostrado ser eficaz para puntuar porciones cortas de una secuencia codificante (CDS) aisladamente de la secuencia completa, lo cual es necesario cuando se analizan microproteínas o exones individuales de genes codificantes de proteínas estándar48,49.
En particular, la reciente integración de los centros de seguimiento de PhyloCSF en el Genome Browser 49,50,51 de la Universidad de California en Santa Cruz (UCSC) permite a los investigadores de todos los orígenes acceder fácilmente a una interfaz fácil de usar para consultar las regiones genómicas de interés para el potencial de codificación de proteínas. El protocolo que se describe a continuación proporciona instrucciones detalladas sobre cómo cargar los centros de seguimiento de PhyloCSF en el UcSC Genome Browser y, posteriormente, interrogar las regiones genómicas de interés para sondear las regiones codificantes de proteínas de alta confianza (o la falta de ellas). Además, en el caso de que se observe una puntuación positiva de PhyloCSF, se delinean pasos para analizar más a fondo el potencial de codificación de microproteínas y generar de manera eficiente alineaciones de múltiples especies de las secuencias de aminoácidos identificadas para ilustrar la conservación de secuencias entre especies. Por último, en la discusión se introducen varios recursos y herramientas adicionales disponibles públicamente para estudiar las características de las microproteínas identificadas, incluidas las estructuras de dominio predichas y la comprensión de la supuesta función de las microproteínas.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
El protocolo que se describe a continuación detalla los pasos para cargar y navegar por las pistas del navegador PhyloCSF en el UCSC Genome Browser (generado por Mudge et al.49). Para preguntas generales sobre el UCSC Genome Browser, se puede encontrar una extensa Guía del usuario de Genome Browser aquí: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Carga del PhyloCSF Track Hub en el ucsc genome browser
- Abra una ventana del navegador de Internet y navegue hasta el UCSC Genome Browser (https://genome.ucsc.edu/).
- En el encabezado Nuestras herramientas , seleccione la opción Centros de seguimiento .
NOTA: La opción Centros de seguimiento también se puede encontrar en la pestaña Mis datos . - En la pestaña Centros públicos , escriba PhyloCSF en el cuadro Términos de búsqueda . Haga clic en el botón Buscar centros públicos .
- Conéctese a PhyloCSF haciendo clic en el botón Conectar para el nombre del concentrador PhyloCSF (Descripción: Potencial evolutivo de codificación de proteínas medido por PhyloCSF).
NOTA: Este track hub se cargará en numerosos conjuntos, incluidos humanos (hg19 y hg38) y ratones (mm10 y mm39). - Después de hacer clic en conectar, espere a ser redirigido a la página UCSC Genome Browser Gateway (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Navegación a genes de interés utilizando identificadores de genes
- Seleccione la especie y el ensamblaje del genoma que desea consultar. Para consultar una especie diferente (por ejemplo, ratón), seleccione la especie de interés en el encabezado Examinar/Seleccionar especies haciendo clic en el icono correspondiente, o escriba la especie en el cuadro de texto que dice: Introduzca especies, nombre común o ID de conjunto.
NOTA: El conjunto aparece directamente en el encabezado Buscar posición . Normalmente, el valor predeterminado es la Asamblea Humana (por ejemplo, diciembre de 2009 [GRCh37/hg19]). - Elija el conjunto que desea buscar en el encabezado Buscar posición mediante el menú desplegable.
- Ingrese la posición, el símbolo del gen o los términos de búsqueda en el cuadro Posición / Término de búsqueda y haga clic en Ir para navegar a un gen de interés en el Navegador del genoma.
- Si la búsqueda dio lugar a varias coincidencias, espere a ser redirigido a una página que requiera la selección de una posición de interés. Haga clic en el gen apropiado de interés.
3. Navegar a las regiones genómicas de interés utilizando información de secuencia
- Navegue hasta el UCSC Genome Browser (https://genome.ucsc.edu/) y seleccione la herramienta de alineación blast-like (BLAT) en el encabezado Nuestras herramientas para consultar una secuencia específica de ADN o proteína. Alternativamente, coloque el cursor sobre la pestaña Herramientas y seleccione la opción Blat o siga este enlace: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Seleccione la especie (Genoma) y el Ensamblaje de interés utilizando los menús desplegables.
- Defina el tipo de consulta mediante el menú desplegable.
- Pegue la secuencia de interés en el cuadro de texto Genoma de búsqueda blaT y haga clic en Enviar.
- Haga clic en el enlace del navegador debajo del encabezado ACCIONES para navegar a la región genómica de interés.
4. Identificación de sORF conservados utilizando datos de seguimiento de PhyloCSF
- Escanee visualmente el área genómica de interés para calificar positivamente las regiones PhyloCSF (Figura 1).
NOTA: Para obtener una explicación detallada de cómo interpretar visualmente las puntuaciones de PhyloCSF en el UCSC Genome Browser, consulte la sección de resultados representativos a continuación. - Utilice la función de zoom para ampliar las regiones de interés para examinar las características de la secuencia y buscar codones de inicio/parada. Para acercar manualmente, mantenga presionada la tecla Mayús y mantenga presionado el botón del mouse mientras arrastra a lo largo de la región de interés. Alternativamente, use los botones de acercar y alejar en la parte superior de la página para navegar (las opciones de zoom 1.5x, 3x, 10x o base están disponibles).
NOTA: Antes de usar los botones de zoom in/zoom out , es necesario reposicionar el gen para que la región de interés esté en el centro de la pantalla. Para realizar esta acción, haga clic en la imagen y arrástrela hacia la izquierda o hacia la derecha para mover la región genómica horizontalmente como desee o use las flechas de movimiento en la parte superior de la página. - Amplíe el zoom hasta que la secuencia de nucleótidos (base) sea visible.
NOTA: La secuencia de nucleótidos aparecerá directamente encima de la puntuación +1 Smoothed PhyloCSF. - Escanee visualmente la secuencia de nucleótidos cerca del principio y el final de las regiones PhyloCSF con puntuación positiva para identificar los codones de inicio y parada putativos (TGA / TAA / TAG).
NOTA: Si el gen de interés está en la cadena menos de ADN, los codones de inicio y parada serán el complemento inverso (es decir, CAT para el codón de inicio y TCA / TTA / CTA para el codón de parada).
5. Visualización de regiones homólogas en otros genomas
- Pase el mouse sobre el encabezado Ver en la parte superior de la página y haga clic en la opción En otros genomas (Convertir ).
- Defina el genoma de interés utilizando el menú desplegable debajo del encabezado Nuevo genoma .
- Seleccione el conjunto genómico de interés mediante el menú desplegable situado en el encabezado Nuevo conjunto y , a continuación, haga clic en el botón Enviar .
- Una vez que el navegador devuelva una lista de regiones en el nuevo ensamblado con similitud, haga clic en el enlace de posición cromosómica para navegar a la región homóloga de interés.
NOTA: El porcentaje de bases totales (nucleótidos) y el lapso que cubre la región se definirán para cada región enumerada. Cuanto mayor sea el porcentaje de bases coincidentes, mayor será la conservación para la región de interés. - Siga las mismas estrategias de navegación detalladas en la Sección 4 para analizar la secuencia.
6. Generación de alineaciones de secuencias multiespecies para microproteínas de interés
- Haga clic en el gen de interés en la pista GENCODE en el UCSC Genome Browser (indicado en la Figura 1A con un cuadro azul) para navegar a la página de descripción del gen.
- En el encabezado Secuencia y enlaces a herramientas y bases de datos , haga clic en el enlace de la tabla que dice Otras especies FASTA.
- Haga clic en las casillas asociadas a las especies de interés para seleccionarlas. Haga clic en Enviar. Copie y pegue las secuencias que aparecen en la parte inferior de la página en formato FASTA en un documento de procesamiento de textos.
- Abra una segunda ventana del navegador y navegue hasta la herramienta Clustal Omega Multiple Sequence Alignment 52 en el sitio web del Instituto Europeo de Bioinformática (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Pegue los archivos de secuencia que todavía están en el portapapeles en el cuadro del STEP 1 que lee secuencias en cualquier formato compatible. Desplácese hasta la parte inferior de la página y haga clic en Enviar. Mire debajo de los resultados alineados (en fuente negra) los símbolos que indican el grado de conservación de cada aminoácido (los símbolos se definen en la Tabla 1).
NOTA: Puede tardar varios minutos en generar la alineación. - Para ver las propiedades de los aminoácidos en color, haga clic en el enlace Mostrar colores directamente encima de las secuencias para colorear los aminoácidos de acuerdo con sus propiedades (definidas en la Tabla 2).
- Copie y pegue la alineación de la secuencia en un programa de procesamiento de textos o presentación de diapositivas para generar una figura o un archivo de ilustración (por ejemplo, la Figura 2).
NOTA: Utilice una fuente monoespaciada para la alineación, como Courier. - Para ver otros resultados de la página de resultados de Clustal Omega , haga clic en las pestañas correspondientes (es decir, Árbol guía o Árbol filogenético).
- Haga clic en la pestaña Visores de resultados para ver las opciones para ver la información de la secuencia utilizando Jalview, un programa gratuito que se especializa en la edición, visualización y análisis de alineación de secuencias múltiples55, o para acceder a enlaces directos a MView y Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Aquí usaremos la microproteína mitoregulina validada (Mtln) como ejemplo para demostrar cómo un sORF conservado generará una puntuación PhyloCSF positiva que se puede visualizar y analizar fácilmente en el UCSC Genome Browser. La mitoregulina se anotó previamente como un ARN no codificante (anteriormente ID del gen humano LINC00116 y ID del gen de ratón 1500011K16Rik). La genómica comparativa y los métodos de análisis de conservación de secuencias desempeñaron un papel crítico en su descubrimiento inicial 40,57,58,59,60,61, destacando la fortaleza de estos métodos. Para este ejemplo, se utilizará el conjunto GRCm38/mm10 del ratón (diciembre de 2011). La búsqueda se puede realizar utilizando los identificadores de genes (mitoregulina, Mtln) o la posición del gen (chr2:127,791,364-127,792,496) como se describe en la sección 2 del protocolo. Alternativamente, la secuencia de aminoácidos para la mitoregulina (que se muestra en la Figura 2) se puede buscar utilizando la herramienta BLAT (descrita en la sección 3 del protocolo).
Aparecerá una pantalla similar a la que se muestra en la Figura 1A con el PhyloCSF Track Hub visible en la parte superior de la pantalla. Las pistas de PhyloCSF suavizadas (suavizadas con un modelo de Markov oculto que define una probabilidad de que cada codón esté codificando) se representan como seis pistas totales, con tres pistas correspondientes a la hebra más de ADN (representada en verde como PhyloCSF +1, +2 y +3) y tres pistas correspondientes a la hebra menos de ADN (representada en rojo como PhyloCSF -1, -2 y -3). Estas pistas representan los tres marcos de lectura potenciales para el gen de interés en cada dirección. En la ventana del navegador, los exones se representan como rectángulos azules conectados por delgadas líneas horizontales azules, que representan los intrones. Las puntas de flecha en las regiones intrónicas indican en qué dirección se transcribe el gen (y, por lo tanto, en qué hebra enfocarse para la puntuación PhyloCSF). Para el ejemplo de Mtln en la Figura 1, las puntas de flecha intrónicas apuntan a la izquierda. Por lo tanto, el gen Mtln se transcribe de la hebra menos de ADN, y la puntuación PhyloCSF relevante se representa en las pistas -1, -2 y -3 (en rojo).
Cada pista de PhyloCSF se representa como una delgada línea negra con regiones de puntuación negativas representadas en verde claro / rojo debajo de la línea y regiones de puntuación positivas indicadas en verde oscuro / rojo sobre la línea. Como se describe en la introducción, una puntuación positiva de PhyloCSF indica una región conservada que probablemente esté codificando. Tenga en cuenta que para las regiones codificantes de proteínas con una conservación de secuencia particularmente alta, a menudo también obtienen una puntuación positiva en la hebra antisentido; sin embargo, la puntuación de PhyloCSF suele ser mayor en la hebra correcta. Por ejemplo, esto se puede ver en la Figura 1 para Mtln, donde la secuencia de codificación correcta puntúa muy alto en la pista PhyloCSF -1, y la hebra antisentido (pista PhyloCSF +2) también genera una puntuación positiva. Como se ve en la Figura 1A (indicada con caja negra), hay una región en el primer exón de Mtln que puntúa muy alto en la pista PhyloCSF -1, lo que sugiere que esto puede corresponder a una región codificante. Para examinar esta región con más detalle, es útil acercar y ampliar la región (Figura 1B). Como se muestra en la Figura 1C,D, la región de puntuación positiva en el primer exón de Mtln comienza directamente sobre un codón de inicio (Figura 1C) y termina en un codón de parada (Figura 1D), lo que indica que este ORF está altamente conservado y sugiere fuertemente que es un ORF codificante. Como Mtln está en la hebra menos de ADN, los codones de inicio y parada se muestran como el complemento inverso del codón (es decir, el codón de inicio ATG se muestra como CAT [Figura 1C] y el codón de parada TGA se muestra como TCA [Figura 1D]).
Además de utilizar PhyloCSF para buscar regiones conservadas con potencial de codificación de microproteínas, esta técnica también se puede aplicar como un análisis de primer paso de RNAs supuestamente no codificantes para descartar la presencia de un ORF conservado, proporcionando así soporte para una anotación no codificante. Por ejemplo, el análisis del lncRNA hotair62,63 bien caracterizado utilizando PhyloCSF muestra una puntuación negativa en todo el gen en las seis pistas (Figura 3), lo que indica fuertemente una falta de conservación de la secuencia y proporciona apoyo de que HOTAIR está correctamente anotado como un ARN no codificante.
Como se ve claramente en la Figura 1, toda la codificación ORF para la mitoregulina se encuentra dentro de un solo exón, produciendo así una lectura simple y directa por parte de PhyloCSF con una sola región, ininterrumpida y de puntuación positiva. Sin embargo, los datos del concentrador de seguimiento de PhyloCSF no siempre son tan claros y fáciles de interpretar. Por ejemplo, la microproteína mitolamban/Stmp1/Mm47 codificada por el gen 1810058I24Rik del ratón 47,64,65 representa un ORF conservado que abarca tres exones (Figura 4A), y la puntuación positiva de PhyloCSF salta de la pista +2 en el exón 1 (Figura 4B) a la pista +3 en el exón 2 (Figura 4C), y luego de vuelta a la pista +2 en el exón 3 (Figura 4D ). Si bien a primera vista esto parece confuso, la explicación es bastante sencilla. PhyloCSF puntúa los seis marcos de lectura potenciales (tres en la cadena más de ADN y tres en la cadena menos) de las regiones genómicas sin considerar la arquitectura específica de exón / intrón para cada gen. Por lo tanto, retiene la información de la secuencia intrónica en la periodicidad de 3 nucleótidos de los marcos de lectura. Por lo tanto, si un intrón contiene un número de nucleótidos que no es divisible por tres (es decir, tres nucleótidos / codón), el marco de lectura PhyloCSF saltará de una pista a otra.
Por último, PhyloCSF también se puede utilizar eficazmente para identificar múltiples ORF codificantes distintos dentro de una sola molécula de ARN. Por ejemplo, la microproteína MIEF1 (MIEF1-MP) está codificada dentro de la UTR 5' del factor de elongación mitocondrial 1 (MIEF1)66 (Figura 5). Cuando la región genómica MIEF1 es analizada por PhyloCSF, una puntuación PhyloCSF positiva discreta correspondiente al MIEF1-MP (Figura 5C) se puede observar fácilmente aguas arriba del CDS principal para MIEF1 (Figura 5B). A continuación se proporciona una discusión adicional sobre MIEF1 y su microproteína asociada (MIEF1-MP) en la discusión junto con un resumen de las fortalezas y debilidades de los métodos y protocolos descritos en este artículo.

Figura 1: El análisis PhyloCSF del gen de la mitoregulina (Mtln) indica una región de conservación de alta secuencia correspondiente a una microproteína validada. (A) Las capturas de pantalla del UCSC Genome Browser y PhyloCSF Tracks muestran que Mtln contiene dos exones y un solo intrón. Las puntas de flecha dentro del intrón apuntan a la izquierda, lo que indica que el gen Mtln se transcribe de la hebra menos de ADN, y las puntuaciones relevantes de PhyloCSF se muestran en las pistas -1, -2 y -3 (en rojo). La secuencia codificante completa de la mitoregulina está contenida dentro del Exón 1 y tiene una puntuación alta en la pista PhyloCSF -1 (B). Un codón de inicio conservado se puede observar claramente al comienzo de la región de puntuación positiva en la pista PhyloCSF -1 (C), que se resalta con un cuadro verde (CAT, complemento inverso ATG). Además, se indica un codón de parada conservado (TCA, complemento inverso TGA) con un cuadro rojo en el panel (D), que se alinea con el final de la región PhyloCSF con puntuación positiva. Puede encontrar información detallada sobre el gen Mtln haciendo clic en el identificador del gen Mtln dentro del cuadro azul (que se muestra en el panel A). Cabe destacar que las regiones codificantes de proteínas altamente conservadas a menudo también obtienen una puntuación positiva en la hebra antisentido (vista aquí en la pista PhyloCSF +2 para Mtln). Sin embargo, la puntuación de PhyloCSF suele ser mayor en la hebra correcta (la pista PhyloCSF -1 en este ejemplo). Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Alineación de secuencias de múltiples especies de la microproteína mitoregulina generada mediante el programa Clustal Omega. Las secuencias de aminoácidos de mitoregulina para las ocho especies indicadas se extrajeron como se detalla en la sección 6 del protocolo y se alinearon con la herramienta de alineación de secuencias múltiples Clustal Omega. Las propiedades de los aminoácidos están indicadas por color (rojo, pequeño/hidrófobo; azul, ácido; magenta, básico; verde, hidroxil/sulfhidrilo/amina) (definido más adelante en la Tabla 2). Los símbolos debajo de los aminoácidos indican el grado de conservación (asteriscos, residuos totalmente conservados; colones, aminoácidos con propiedades muy similares; períodos, conservación entre grupos de propiedades débilmente similares) (detallado ampliamente en la Tabla 1). Haga clic aquí para ver una versión más grande de esta figura.
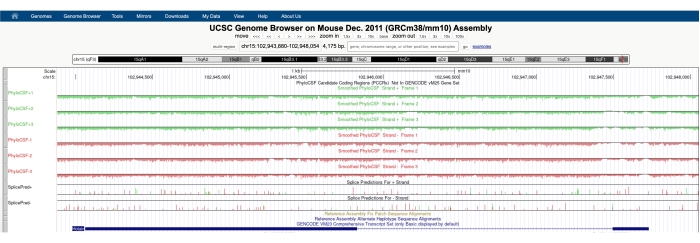
Figura 3: Una captura de pantalla de las pistas de PhyloCSF para el ARN largo no codificante validado Hotair muestra una falta de conservación de la secuencia en todo su locus genómico. Las puntas de flecha en la región intrónica de Hotair apuntan a la izquierda, lo que indica que el lncRNA se transcribe de la cadena negativa de ADN y, por lo tanto, las pistas PhyloCSF -1, -2 y -3 deben ser el foco del análisis. Tenga en cuenta que la puntuación de PhyloCSF es negativa en todo el gen (para las seis pistas), lo que indica una falta de conservación de la secuencia, lo que respalda su anotación adecuada como un ARN no codificante. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Análisis PhyloCSF del gen ratón 1810058I24Rik , que codifica la microproteína mitolamban/Stmp1/Mm47. (A) El gen 1810058I24Rik del ratón está compuesto por tres exones, y las puntas de flecha en las regiones intrónicas apuntan a la derecha, lo que indica que se transcribe en la cadena más de ADN y, por lo tanto, se deben analizar las pistas PhyloCSF +1, +2 y +3. La secuencia codificante de microproteínas conservadas abarca los tres exones, comenzando en el exón 1 (B), leyendo a través del exón 2 (C) y terminando en el exón 3 (D). Tenga en cuenta que la puntuación positiva de PhyloCSF se encuentra en la pista +2 en el exón 1, la pista +3 en el exón 2 y la pista +2 en el exón 1. La razón del movimiento de la puntuación positiva de una pista a la otra es que PhyloCSF analiza los seis marcos de lectura potenciales de la secuencia de ADN independientemente de la estructura exón/intrón del gen. Por lo tanto, un intrón que contiene un número de nucleótidos que no es divisible por tres (tres nucleótidos/codón) causará un cambio en el marco de lectura a una pista diferente. Haga clic aquí para ver una versión más grande de esta figura.

Figura 5: Análisis del locus genómico Mief1 con PhyloCSF identifica una región con potencial de codificación de proteínas en el UTR 5' que es independiente del CDS Mief1 principal en el ARN compartido. Se ha demostrado que este ORF aguas arriba conservado (uORF) codifica una microproteína llamada Mief1-MP. (A) Visión general del locus genómico Mief1 . Las puntas de flecha en los intrones apuntan a la derecha, lo que indica que Mief1 se transcribe de la hebra más de ADN (concéntrese en las pistas PhyloCSF +1, +2 y +3 para determinar el potencial de codificación). El CDS principal de Mief1 codifica una proteína de 463 aminoácidos y se muestra en el panel (B). Sin embargo, también hay un ORF aguas arriba conservado distinto dentro de los 5' UTR de Mief1 que codifica una microproteína única de 70 aminoácidos llamada Mief1-MP (C). Como se ve en el Panel C, el Mief1-MP tiene su propio codón de inicio y parada conservado dentro del UtR Mief1 5', y el ORF obtiene una puntuación muy alta en la pista PhyloCSF +1, proporcionando una fuerte evidencia de que codifica una microproteína funcional. Abreviaturas: ORF = marco de lectura abierto; uORF = ORF ascendente; UTR = región no traducida; CDS = secuencia de codificación. Haga clic aquí para ver una versión más grande de esta figura.
| Símbolo | Nivel de conservación de aminoácidos | Aminoácidos agrupados |
| Asterisco (*) | Residuo totalmente conservado | No aplicable (residuo único y totalmente conservado) |
| Colon (:) | Grupos con propiedades muy similares | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW (en inglés) |
| Periodo (.) | Grupos con propiedades débilmente similares | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY |
| Espacio (sin símbolo) | Sin similitud | No aplicable (sin similitud) |
Tabla 1: Definiciones de símbolos de consenso para Alineaciones de Secuencia Múltiple generadas por Clustal Omega. La alineación de la secuencia de múltiples especies que se muestra en la Figura 2 se generó utilizando Clustal Omega52. Abreviaturas: serina (S), treonina (T), alanina (A), asparagina (N), ácido glutámico (E), glutamina (Q), lisina (K), ácido aspártico (D), arginina (R), metionina (M), isoleucina (I), leucina (L), fenilalanina (F), histidina (H), tirosina (Y), triptófano (W), cisteína (C), valina (V), glicina (G), prolina (P).
| Color de fuente | Propiedad | Residuo de aminoácidos [Abreviatura] |
| Rojo | Pequeño, hidrófobo | alanina [A], valina [V], fenilalanina [F], prolina [P], metionina [M], isoleucina [I], leucina [L], triptófano [W] |
| Azul | Ácido | ácido aspártico [D], ácido glutámico [E] |
| Magenta | Básico | arginina [R], lisina [K] |
| Verde | Hidroxl, sulfhidrilo, amina, +G | serina [S], treonina [T], tirosina [Y], histidina [H], cisteína [C], asparagina [N], glicina [G], glutamina [Q] |
Tabla 2: Propiedades de los aminoácidos representados en la Figura 2. Se utilizó Clustal Omega52 para generar la alineación de secuencias múltiples que se muestra en la Figura 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
El protocolo presentado aquí proporciona instrucciones detalladas sobre cómo interrogar las regiones genómicas de interés para el potencial de codificación de microproteínas utilizando PhyloCSF en el navegador del genoma UCSC fácil de usar 48,49,50,51. Como se detalló anteriormente, PhyloCSF es un poderoso algoritmo de genómica comparativa que integra modelos filogenéticos y frecuencias de sustitución de codones para identificar firmas evolutivas que son típicas de los genes codificantes de proteínas48,49. PhyloCSF se ha utilizado ampliamente para identificar microproteínas funcionales en regiones genómicas previamente anotadas como no codificantes 38,39,40,41,42,43,44,45,46,47 , y se ha demostrado que este enfoque supera a otros métodos de genómica comparativa para secuencias cortas como microproteínas tan pequeñas como 13 aminoácidos y para pequeños exones de proteínas canónicas 35,48,49. En particular, la utilidad de PhyloCSF como un método robusto para identificar secuencias funcionales de codificación de proteínas a través de la conservación evolutiva se extiende más allá de la de las especies de vertebrados e invertebrados e incluso se ha aplicado recientemente a los genomas virales para interrogar con éxito la capacidad de codificación de proteínas del genoma del SARS-CoV-267.
Además de identificar secuencias de codificación putativas dentro de ARN no codificantes anotados, una ventaja de PhyloCSF es que también puede detectar de manera confiable microproteínas conservadas codificadas por ORF dentro de regiones no traducidas anotadas (UTR) de genes codificantes de proteínas canónicas, incluidos orfs 5' aguas arriba y 3' aguas abajo (uORFs y dORFs, respectivamente)8,19,66,68 . Por ejemplo, la microproteína MIEF1 (MIEF1-MP) está codificada en la UTR 5' del factor de elongación mitocondrial 1 (MIEF1)66. En el caso de MIEF1-MP, se observa una puntuación PhyloCSF positiva discreta correspondiente al MIEF1-MP aguas arriba del ORF que codifica MIEF1 (Figura 5). Mientras que algunas microproteínas codificadas uORF interactúan directamente con las proteínas canónicas aguas abajo en su ARNm compartido, (por ejemplo, MIEF1-MP y MIEF1), otras funcionan independientemente de la proteína codificada por el CDS principal66,68. Por lo tanto, al caracterizar microproteínas codificadas por uORF, no se debe asumir que funcionan a través de la regulación directa de su producto proteico aguas abajo.
Si bien PhyloCSF tiene muchas fortalezas claras como herramienta para la identificación de secuencias codificantes de microproteínas conservadas, es importante reconocer varias limitaciones de este método. En primer lugar, si bien la conservación de la secuencia sugiere fuertemente que una región genómica ha sido sometida a selección funcional y, por lo tanto, está codificando, la falta de conservación robusta y una puntuación PhyloCSF negativa resultante no descartan definitivamente el potencial de codificación para una secuencia dada. En otras palabras, confiar exclusivamente en PhyloCSF puede resultar en la supervisión de ORF traducidos que no están fuertemente conservados pero que aún producen microproteínas funcionales. En particular, las regiones genómicas con puntuaciones de conservación bajas o negativas de conservación podrían corresponder a regiones codificantes específicas de la especie o a las de genes evolutivos "jóvenes" a través de la divergencia de secuencia o el nacimiento de novo del gen 46,69,70,71,72,73,74. Por ejemplo, la microproteína ASAP, que está codificada por lo que antes se pensaba que era el ARN humano no codificante LINC00467, no es calificada positivamente por PhyloCSF porque la secuencia de aminoácidos solo se conserva en mamíferos superiores75. Además, estudios recientes identificaron varias microproteínas específicas para humanos, incluida una codificada por el lncRNA intergénico RP3-527G5.1, que no genera una puntuación positiva de PhyloCSF68,72. En este sentido, la ausencia de una puntuación positiva de PhyloCSF no puede interpretarse como prueba de una región no codificante y debe interpretarse con precaución.
Una segunda consideración a tener en cuenta al usar PhyloCSF es que a pesar de que una puntuación positiva es altamente sugestiva de selección funcional y capacidad de codificación de proteínas, esta línea de evidencia no puede ser independiente y debe ser validada experimentalmente. Ejemplos de métodos que se pueden utilizar para generar evidencia de apoyo para la expresión estable de microproteínas incluyen la detección de la proteína putativa por espectrometría de masas o western blotting utilizando un anticuerpo levantado contra la secuencia de microproteínas de interés. Alternativamente, dado que puede ser difícil generar anticuerpos confiables para microproteínas debido a la falta de opciones de secuencia para una antigenicidad óptima, también es posible usar CRISPR / Cas9 y la vía de reparación dirigida por homología (HDR) para introducir una etiqueta de epítopo en el locus endógeno en marco con la secuencia de microproteínas putativa, facilitando así la detección de la proteína de interés utilizando un anticuerpo de alta afinidad (por ejemplo, BANDERA, HA, V5, Myc)18. Una limitación final de PhyloCSF para reconocer es que aunque actualmente está integrado en muchos de los ensamblajes genómicos comúnmente utilizados, incluidos Homo sapiens (humano hg19, hg38), Mus musculus (ratón mm10, mm39), Gallus gallus (pollo, galGal4, galGal6), Drosophila melanogaster (mosca de la fruta, dm6), Caenorhabditis elegans (nematodos, ce11) y SARS-CoV-2 (wuhCor1), todavía hay muchas especies que actualmente no se pueden consultar directamente en el UCSC Genome Browser.
La identificación de dominios conservados o características de secuencia dentro de las microproteínas identificadas puede ayudar a aumentar la confianza en su relevancia funcional y proporcionar una idea de su función putativa. Aquí proporcionamos recomendaciones para herramientas y recursos específicos que se pueden utilizar para analizar las secuencias de aminoácidos de microproteínas identificadas con más detalle para obtener dicha información. Las herramientas específicas que se enumeran a continuación (y se resumen en la Tabla de Materiales) están disponibles gratuitamente para el público, y hemos encontrado que son particularmente fáciles de usar y robustas en estudios de microproteínas 18,38,39,40,41,47. Más allá de las herramientas descritas aquí, hay una multitud de recursos adicionales que se pueden encontrar en portales de recursos bioinformáticos como Expasy (https://www.expasy.org) y EMBL-EBI (https://www.ebi.ac.uk/services/all). Sin embargo, detallar los detalles específicos de cada una de las herramientas dentro de estos repositorios está más allá del alcance de este artículo. Aquí te recomendamos los siguientes recursos.
En primer lugar, TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) analiza secuencias de proteínas de interés para detectar la presencia de dominios transmembrana. En particular, una serie de microproteínas que se han caracterizado funcionalmente hasta ahora contienen dominios transmembrana de un solo paso, lo que facilita su localización en las regiones de membrana y permite su regulación directa de los canales iónicos, intercambiadores y enzimas asociadas a la membrana30. En segundo lugar, el Centro Nacional de Información Biotecnológica (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) es una herramienta popular utilizada para identificar dominios conservados dentro de secuencias de proteínas o nucleótidos codificantes. En tercer lugar, la base de datos de la familia de proteínas (Pfam)78 (http://pfam.xfam.org) proporciona alineaciones y clasificaciones de familias y dominios de proteínas. En cuarto lugar, WoLF PSORT79 (https://wolfpsort.hgc.jp/) es una herramienta que se puede emplear para predecir la localización de proteínas subcelulares. En quinto lugar, COXPRESdB80 es una base de datos de coexpresión génica (https://coxpresdb.jp) que proporciona relaciones genéticas correguladas para estimar las funciones génicas. Finalmente, SignalP 6.081 es un programa de predicción ampliamente utilizado (https://services.healthtech.dtu.dk/service.php?SignalP) que reconoce la presencia de una secuencia de péptidos de señal y predice la ubicación del sitio de escisión.
En resumen, los métodos descritos aquí se pueden utilizar para analizar eficazmente las regiones genómicas de interés para el potencial de codificación de proteínas utilizando PhyloCSF en el UCSC Genome Browser. Estos métodos son altamente accesibles y pueden ser fácilmente aprendidos y aplicados de manera eficiente por individuos sin capacitación previa o experiencia en bioinformática o genómica comparativa. Como se demuestra aquí en detalle, PhyloCSF es una herramienta poderosa que se puede aplicar como un análisis de primer paso para ayudar a distinguir los genes codificantes de proteínas frente a los no codificantes en los genomas de vertebrados, invertebrados y virales, y las fortalezas de este enfoque superan en gran medida las debilidades observadas.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores declaran que no tienen intereses financieros contrapuestos.
Acknowledgments
Este trabajo fue apoyado por subvenciones de los Institutos Nacionales de Salud (HL-141630 y HL-160569) y la Fundación de Investigación Infantil de Cincinnati (Premio Del Fideicomisario).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
