

Summary
Протокол, описанный здесь, предоставляет подробные инструкции о том, как анализировать геномные области, представляющие интерес, на потенциал кодирования микропротеинов с помощью PhyloCSF в удобном для пользователя браузере генома UCSC. Кроме того, рекомендуется несколько инструментов и ресурсов для дальнейшего изучения характеристик последовательности идентифицированных микропротеинов, чтобы получить представление об их предполагаемых функциях.
Abstract
Секвенирование следующего поколения (NGS) продвинуло область геномики вперед и произвело целые последовательности генома для многочисленных видов животных и модельных организмов. Однако, несмотря на это богатство информации о последовательностях, комплексные усилия по аннотированию генов оказались сложными, особенно для небольших белков. Примечательно, что обычные методы аннотирования белков были разработаны для преднамеренного исключения предполагаемых белков, кодируемых короткими открытыми кадрами считывания (sORFs) длиной менее 300 нуклеотидов, чтобы отфильтровать экспоненциально большее количество ложных некодирующих SORF по всему геному. В результате сотни функциональных небольших белков, называемых микропротеинами (длиной <100 аминокислот), были неправильно классифицированы как некодирующие РНК или полностью упущены из виду.
Здесь мы предоставляем подробный протокол для использования бесплатных, общедоступных биоинформационных инструментов для запроса геномных областей на наличие потенциала кодирования микропротеинов на основе эволюционного сохранения. В частности, мы предоставляем пошаговые инструкции о том, как исследовать потенциал сохранения последовательностей и кодирования с использованием филогенетических частот замещения кодонов (PhyloCSF) в удобном для пользователя браузере генома Калифорнийского университета Санта-Крус (UCSC). Кроме того, мы подробно описываем шаги по эффективному созданию множественных видовых выравниваний идентифицированных последовательностей микропротеинов для визуализации сохранения аминокислотных последовательностей и рекомендуем ресурсы для анализа характеристик микропротеинов, включая прогнозируемые доменные структуры. Эти мощные инструменты могут быть использованы, чтобы помочь идентифицировать предполагаемые микропротеин-кодирующие последовательности в неканонических геномных областях или исключить наличие сохраненной кодирующей последовательности с трансляционным потенциалом в некодирующей транскрипте, представляющей интерес.
Introduction
Идентификация полного набора кодирующих элементов в геноме была основной целью с момента начала проекта «Геном человека» и остается центральной задачей для понимания биологических систем и этиологии генетических заболеваний 1,2,3,4. Достижения в методах NGS привели к производству целых последовательностей генома для широкого круга организмов, включая позвоночных, беспозвоночных, дрожжи и растения5. Кроме того, высокопроизводительные методы транскрипционного секвенирования дополнительно выявили сложность клеточного транскриптома и идентифицировали тысячи новых молекул РНК с кодирующими белки и некодирующими функциями 6,7. Расшифровка этого огромного количества информации о последовательностях является непрерывным процессом, и проблемы остаются с комплексными усилиями по аннотированию генов8.
Недавняя разработка методов трансляционного профилирования, включая профилирование рибосом 9,10 и секвенирование полирибосом11, предоставила доказательства, указывающие на то, что сотни неканонических событий трансляции сопоставляются с неаннотированными в настоящее время sORFs по всему геному, с потенциалом генерировать небольшие белки, называемые микропротеинами или микропептидами 12,13,14,15,16. 17. Микропротеины появились как новый класс универсальных белков, ранее упускавшихся из виду стандартными методами аннотирования генов из-за их небольшого размера (<100 аминокислот) и отсутствия классических кодирующих белок характеристик генов 8,12,18,19,20. Микропротеины были описаны практически во всех организмах, включая дрожжи21,22, мух 17,23,24 и млекопитающих 25,26,27,28, и было показано, что они играют решающую роль в различных процессах, включая развитие, метаболизм и передачу сигналов о стрессе 19,20,29. 30,31,32,33,34. Таким образом, крайне важно продолжать добывать геном для дополнительных членов этого давно забытого класса функциональных малых белков.
Несмотря на широкое признание биологической важности микропротеинов, этот класс генов остается значительно недопредставленным в аннотациях генома, и их точная идентификация по-прежнему является постоянной проблемой, которая препятствует прогрессу в этой области. Недавно были разработаны различные вычислительные инструменты и экспериментальные методы для преодоления трудностей, связанных с идентификацией микропротеин-кодирующих последовательностей (подробно обсуждается в нескольких всеобъемлющих обзорах 8,35,36,37). Многие недавние исследования по идентификации микропротеинов 38,39,40,41,42,43,44,45,46,47 в значительной степени опирались на использование одного такого алгоритма под названием PhyloCSF48,49 , мощный подход к сравнительной геномике, который может быть использован для отличия сохраненных белково-кодирующих областей генома от тех, которые не являются кодирующими.
PhyloCSF сравнивает частоты замещения кодонов (CSF) с использованием многовидовых нуклеотидных выравниваний и филогенетических моделей для обнаружения эволюционных сигнатур генов, кодирующих белок. Этот эмпирический подход, основанный на модели, опирается на предпосылку, что белки в основном сохраняются на уровне аминокислот, а не на нуклеотидной последовательности. Поэтому синонимичные замены кодонов, которые кодируют одну и ту же аминокислоту, или замены кодонов на аминокислоты с сохраненными свойствами (т. е. заряд, гидрофобность, полярность) оцениваются положительно, в то время как несинонимные замены, включая неправильные и бессмысленные замены, оцениваются отрицательно. PhyloCSF обучен на данных всего генома и доказал свою эффективность в оценке коротких участков кодирующей последовательности (CDS) в отрыве от полной последовательности, что необходимо при анализе микропротеинов или отдельных экзонов стандартных белково-кодирующих генов48,49.
Примечательно, что недавняя интеграция трековых хабов PhyloCSF в браузере генома 49,50,51 Калифорнийского университета в Санта-Крус (UCSC) позволяет исследователям всех слоев общества легко получить доступ к удобному интерфейсу для запроса геномных областей, представляющих интерес для потенциала кодирования белка. Протокол, описанный ниже, предоставляет подробную инструкцию о том, как загрузить концентраторы трека PhyloCSF в браузер генома UCSC и впоследствии опрашивать геномные области, представляющие интерес для исследования высоконадежных белково-кодирующих областей (или их отсутствия). Кроме того, в случае, когда наблюдается положительная оценка PhyloCSF, описываются шаги для дальнейшего анализа потенциала кодирования микропротеинов и эффективного создания нескольких видовых выравниваний идентифицированных аминокислотных последовательностей для иллюстрации сохранения межвидовых последовательностей. Наконец, в ходе обсуждения был представлен ряд дополнительных общедоступных ресурсов и инструментов для изучения выявленных характеристик микропротеинов, включая прогнозируемые структуры доменов и понимание предполагаемой функции микропротеинов.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Протокол, описанный ниже, описывает шаги для загрузки и навигации по дорожкам браузера PhyloCSF в браузере генома UCSC (сгенерированном Mudge et al.49). Для общих вопросов, касающихся браузера генома UCSC, обширное руководство пользователя браузера генома можно найти здесь: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Загрузка PhyloCSF Track Hub в браузер генома UCSC
- Откройте окно интернет-браузера и перейдите к браузеру генома UCSC (https://genome.ucsc.edu/).
- Под заголовком Наши инструменты выберите параметр Отслеживать концентраторы .
ПРИМЕЧАНИЕ: Опцию Track Hubs также можно найти на вкладке Мои данные . - На вкладке Общедоступные концентраторы введите PhyloCSF в поле Условия поиска . Нажмите кнопку Поиск в общедоступных концентраторах .
- Подключитесь к PhyloCSF, нажав кнопку «Подключиться » для имени концентратора PhyloCSF (Описание: Эволюционный потенциал кодирования белка, измеренный PhyloCSF).
ПРИМЕЧАНИЕ: Этот Track Hub будет загружаться на многочисленные сборки, включая человека (hg19 и hg38) и мышь (mm10 и mm39). - После нажатия кнопки «Подключиться» подождите, пока вас перенаправят на страницу UCSC Genome Browser Gateway (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Переход к генам, представляющим интерес, с помощью идентификаторов генов
- Выберите вид и сборку генома для запроса. Чтобы запросить другой вид (например, мышью), выберите интересующий вид под заголовком «Обзор/Выбор видов », щелкнув соответствующий значок, или введите вид в текстовое поле с надписью «Введите виды, общее название или идентификатор сборки».
ПРИМЕЧАНИЕ: Сборка указана непосредственно под заголовком Найти позицию . Как правило, по умолчанию используется человеческая сборка (например, декабрь 2009 г. [GRCh37/hg19]). - Выберите сборку для поиска под заголовком Найти позицию с помощью раскрывающегося меню.
- Введите позицию, символ гена или условия поиска в поле «Позиция/Условие поиска» и нажмите « Перейти», чтобы перейти к интересующему гену в браузере генома.
- Если поиск привел к нескольким совпадениям, подождите, пока вас перенаправят на страницу, которая требует выбора интересующей позиции. Нажмите на соответствующий ген, представляющий интерес.
3. Навигация по интересующим геномным областям с использованием информации о последовательностях
- Перейдите в браузер генома UCSC (https://genome.ucsc.edu/) и выберите BLAST-Like Alignment Tool (BLAT) под заголовком Наши инструменты для запроса определенной последовательности ДНК или белка. Кроме того, наведите курсор на вкладку Сервис и выберите опцию Blat или перейдите по этой ссылке: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Выберите интересующий вид (Геном) и Сборку с помощью выпадающих меню.
- Определите тип запроса с помощью раскрывающегося меню.
- Вставьте интересующую последовательность в текстовое поле BLAT Search Genome и нажмите кнопку Отправить.
- Нажмите на ссылку браузера под заголовком ACTIONS , чтобы перейти к интересующей геномной области.
4. Идентификация сохраненных sORF с помощью данных трека PhyloCSF
- Визуально сканируйте интересующую геномную область для положительной оценки областей PhyloCSF (рисунок 1).
ПРИМЕЧАНИЕ: Подробное объяснение того, как визуально интерпретировать оценки PhyloCSF в браузере генома UCSC, см. в разделе репрезентативных результатов ниже. - Используйте функцию масштабирования для увеличения областей, представляющих интерес, для изучения характеристик последовательности и поиска кодонов запуска/остановки. Чтобы увеличить масштаб вручную, удерживайте клавишу Shift, а затем щелкните и удерживайте кнопку мыши, перетаскивая вдоль интересующей области. Кроме того, для навигации можно использовать кнопки увеличения и уменьшения масштаба в верхней части страницы (доступны параметры масштабирования 1,5x, 3x, 10x или base zoom).
ПРИМЕЧАНИЕ: Перед использованием кнопок увеличения/уменьшения масштаба необходимо изменить положение гена так, чтобы интересующая область находилась в середине экрана. Чтобы выполнить это действие, нажмите на изображение и перетащите его влево или вправо, чтобы переместить геномную область горизонтально по желанию, или используйте стрелки перемещения в верхней части страницы. - Увеличивайте масштаб до тех пор, пока не станет видна нуклеотидная (базовая) последовательность.
ПРИМЕЧАНИЕ: Нуклеотидная последовательность появится непосредственно над оценкой +1 Smoothed PhyloCSF. - Визуально сканируйте нуклеотидную последовательность вблизи начала и конца положительно оцениваемых областей PhyloCSF, чтобы идентифицировать предполагаемые начальные (ATG) и остановочные (TGA / TAA / TAG) кодоны.
ПРИМЕЧАНИЕ: Если интересующий ген находится на минусовой цепи ДНК, то стартовый и стоп-кодоны будут обратным дополнением (т.е. CAT для стартового кодона и TCA/TTA/CTA для стоп-кодона).
5. Просмотр гомологичных областей в других геномах
- Наведите указатель мыши на заголовок Вид в верхней части страницы и нажмите на опцию В других геномах (Преобразовать ).
- Определите интересующий геном с помощью раскрывающегося меню под заголовком Новый геном .
- Выберите интересующую геномную сборку с помощью раскрывающегося меню под заголовком Новая сборка , затем нажмите кнопку Отправить .
- Как только браузер вернет список областей в новой сборке со сходством, щелкните ссылку на положение хромосомы , чтобы перейти к гомологичной интересующей области.
ПРИМЕЧАНИЕ: Процент общих оснований (нуклеотидов) и диапазон, охватываемый областью, будут определены для каждого из перечисленных регионов. Чем выше процент соответствующих баз, тем выше сохранение для интересующего региона. - Следуйте тем же навигационным стратегиям, описанным в разделе 4, чтобы проанализировать последовательность.
6. Создание многовидовых выравниваний последовательностей для интересующих микропротеинов
- Нажмите на интересующий ген в треке GENCODE в браузере генома UCSC (показан на рисунке 1A синим прямоугольником), чтобы перейти на страницу описания гена.
- Под заголовком Последовательность и ссылки на инструменты и базы данных нажмите на ссылку в таблице, которая гласит Другие виды FASTA.
- Нажмите на поля, связанные с интересующими видами, чтобы выбрать их. Нажмите кнопку Отправить. Скопируйте и вставьте последовательности, появившиеся внизу страницы в формате FASTA, в текстовый документ.
- Откройте второе окно браузера и перейдите к инструменту Clustal Omega Multiple Sequence Alignment tool 52 на веб-сайте Европейского института биоинформатики (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Вставьте файлы последовательностей, которые все еще находятся в буфере обмена, в поле на шаге 1 , которое считывает последовательности в любом поддерживаемом формате. Прокрутите страницу вниз и нажмите «Отправить». Ниже приведены выровненные результаты (черным шрифтом) для символов, которые указывают на степень сохранности каждой аминокислоты (символы определены в таблице 1).
ПРИМЕЧАНИЕ: Создание выравнивания может занять несколько минут. - Чтобы просмотреть свойства аминокислот в цвете, нажмите на ссылку Показать цвета непосредственно над последовательностями, чтобы окрасить аминокислоты в соответствии с их свойствами (определенными в таблице 2).
- Скопируйте и вставьте выравнивание последовательности в программу обработки текста или слайд-шоу для создания файла рисунка или иллюстрации (например, рисунок 2).
ПРИМЕЧАНИЕ: Используйте моноширинный шрифт для выравнивания, например Courier. - Чтобы просмотреть другие выходные данные со страницы результатов Clustal Omega , нажмите на соответствующие вкладки (например, Guide Tree или Phylogenetic Tree).
- Перейдите на вкладку Средства просмотра результатов , чтобы просмотреть информацию о последовательностях с помощью Jalview, бесплатной программы, которая специализируется на редактировании, визуализации и анализе нескольких последовательностей55, или для доступа к прямым ссылкам на MView и Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Здесь мы будем использовать проверенный микропротеин миторегулин (Mtln) в качестве примера, чтобы продемонстрировать, как законсервированный sORF будет генерировать положительную оценку PhyloCSF, которую можно легко визуализировать и проанализировать в браузере генома UCSC. Миторегулин ранее был аннотирован как некодирующая РНК (ранее человеческий ген ID LINC00116 и мышиный ген ID 1500011K16Rik). Сравнительная геномика и методы анализа сохранения последовательностей сыграли решающую роль в его первоначальном открытии 40,57,58,59,60,61, подчеркнув силу этих методов. В этом примере будет использоваться мышь GRCm38/mm10 (декабрь 2011 г.). Поиск может быть выполнен с использованием идентификаторов генов (mitoregulin, Mtln) или позиции гена (chr2:127,791,364-127,792,496), как описано в разделе 2 протокола. Альтернативно, аминокислотная последовательность для миторегулина (показанная на фиг.2) может быть найдена с помощью инструмента BLAT (описанного в разделе 3 протокола).
Появится экран, похожий на показанный на рисунке 1A , с PhyloCSF Track Hub, видимым в верхней части экрана. Сглаженные треки PhyloCSF (сглаженные скрытой моделью Маркова, определяющей вероятность того, что каждый кодон кодирует) изображены в виде шести общих треков, с тремя дорожками, соответствующими плюсовой нити ДНК (изображены зеленым цветом как PhyloCSF +1, +2 и +3) и тремя дорожками, соответствующими минусовой нити ДНК (изображены красным цветом как PhyloCSF -1, -2 и -3). Эти треки представляют собой три потенциальных кадра считывания для гена, представляющего интерес в каждом направлении. В окне браузера экзоны изображены в виде синих прямоугольников, соединенных тонкими синими горизонтальными линиями, которые представляют собой интроны. Наконечники стрел на интронных областях указывают, в каком направлении транскрибируется ген (и, следовательно, на какой нити сосредоточиться для оценки PhyloCSF). Для примера Mtln на рисунке 1 интронные наконечники стрелок указывают влево. Таким образом, ген Mtln транскрибируется из минусовой нити ДНК, а соответствующая оценка PhyloCSF изображена в треках -1, -2 и -3 (красным цветом).
Каждый трек PhyloCSF изображен в виде тонкой черной линии с отрицательными областями оценки, изображенными светло-зеленым / красным цветом под линией, и положительными областями оценки, обозначенными темно-зеленым / красным над линией. Как описано во введении, положительная оценка PhyloCSF указывает на сохраненную область, которая, вероятно, кодирует. Обратите внимание, что для белково-кодирующих областей с особенно высоким сохранением последовательности они часто также положительно оценивают антисмысловую нить; тем не менее, оценка PhyloCSF обычно выше на правильной нити. Например, это можно увидеть на рисунке 1 для Mtln, где правильная последовательность кодирования имеет очень высокую оценку в треке PhyloCSF -1, а антисмысловая нить (трек PhyloCSF +2) также генерирует положительную оценку. Как видно на рисунке 1A (обозначенном черным ящиком), в первом экзоне Mtln есть область, которая очень высоко оценивается на треке PhyloCSF -1, предполагая, что это может соответствовать кодирующей области. Чтобы изучить эту область более подробно, полезно увеличить и увеличить область (рисунок 1B). Как показано на рисунке 1C,D, положительно оценивающая область в первом экзоне Mtln начинается непосредственно над стартовым кодоном (рисунок 1C) и заканчивается стоп-кодоном (рисунок 1D), что указывает на то, что этот ORF сильно сохраняется и убедительно предполагает, что это кодирующий ORF. Поскольку Mtln находится на минусовой нити ДНК, кодоны запуска и остановки показаны как обратное дополнение кодона (т. Е. Кодон запуска ATG показан как CAT [Рисунок 1C], а стоп-кодон TGA показан как TCA [Рисунок 1D]).
В дополнение к использованию PhyloCSF для поиска законсервированных областей с потенциалом микропротеин-кодирования, этот метод также может быть применен в качестве анализа первого прохода предполагаемых некодирующих РНК, чтобы исключить наличие сохраненного ORF, тем самым обеспечивая поддержку некодирующей аннотации. Например, анализ хорошо охарактеризованной lncRNA HOTAIR62,63 с использованием PhyloCSF показывает отрицательный балл по всему гену по всем шести дорожкам (рисунок 3), что убедительно указывает на отсутствие сохранения последовательности и подтверждает, что HOTAIR правильно аннотирован как некодирующая РНК.
Как ясно видно на рисунке 1, весь кодирующий ORF для миторегулина расположен в пределах одного экзона, тем самым производя простое и понятное считывание PhyloCSF с одной, непрерывной, положительно оценивающей областью. Однако данные концентратора треков PhyloCSF не всегда так четко и легко интерпретируются. Например, микропротеин mitolamban/Stmp1/Mm47, закодированный мышью 1810058I24Rik гена 47,64,65, изображает законсервированный ORF, который охватывает три экзона (рисунок 4A), а положительная оценка PhyloCSF прыгает с трека +2 в экзоне 1 (рисунок 4B) на трек +3 в экзоне 2 (рисунок 4C), а затем обратно к дорожке +2 в экзоне 3 (рисунок 4D). ). Хотя на первый взгляд это выглядит запутанным, объяснение довольно простое. PhyloCSF оценивает шесть потенциальных кадров считывания (три на плюсовой цепи ДНК и три на минусовой цепи) геномных областей без учета конкретной архитектуры экзона/ интрона для каждого гена. Поэтому он сохраняет информацию об интронной последовательности в 3-нуклеотидной периодичности считывающих кадров. Таким образом, если интрон содержит ряд нуклеотидов, которое не делится на три (т.е. три нуклеотида/кодона), кадр считывания PhyloCSF будет перескакивать с одного трека на другой.
Наконец, PhyloCSF также может быть эффективно использован для идентификации нескольких различных кодирующих ORF в одной молекуле РНК. Например, микропротеин MIEF1 (MIEF1-MP) кодируется в пределах 5' UTR фактора удлинения митохондрий 1 (MIEF1)66 (Рисунок 5). Когда геномная область MIEF1 анализируется PhyloCSF, дискретный положительный балл PhyloCSF, соответствующий MIEF1-MP (рисунок 5C), можно легко наблюдать выше по течению основного CDS для MIEF1 (рисунок 5B). Дальнейшее обсуждение ММСО1 и связанных с ним микропротеинов (ММСО1-МП) приводится ниже в ходе обсуждения вместе с кратким изложением сильных и слабых сторон методов и протоколов, изложенных в этой статье.

Рисунок 1: PhyloCSF анализ гена миторегулина (Mtln) указывает на область сохранения высокой последовательности, соответствующую проверенному микропротеину. (A) Скриншоты браузера генома UCSC и треков PhyloCSF показывают, что Mtln содержит два экзона и один интрон. Наконечники стрел внутри интрона указывают на то, что ген Mtln транскрибируется из минусовой нити ДНК, и поэтому соответствующие оценки PhyloCSF показаны в треках -1, -2 и -3 (красным цветом). Полная кодирующая последовательность миторегулина содержится в Exon 1 и имеет высокие оценки на треке PhyloCSF -1 (B). Законсервированный стартовый кодон можно четко наблюдать в начале положительно оценивающей области в треке PhyloCSF -1 (C), который подсвечивается зеленым прямоугольником (CAT, обратное дополнение ATG). Кроме того, законсервированный стоп-кодон (TCA, обратный комплемент TGA) обозначен красным прямоугольником на панели (D), который выравнивается с концом положительно оценивающей области PhyloCSF. Подробную информацию о гене Mtln можно найти, нажав на идентификатор гена Mtln в синем поле (показано на панели A). Следует отметить, что высококонсервативные белковые кодирующие области часто также положительно оценивают антисмысловую нить (см. здесь в треке PhyloCSF +2 для Mtln). Тем не менее, оценка PhyloCSF обычно выше на правильной нити (трек PhyloCSF -1 в этом примере). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 2: Выравнивание последовательностей нескольких видов микропротеина миторегулина, генерируемого с помощью программы Clustal Omega. Последовательности аминокислот миторегулина для восьми указанных видов были извлечены, как описано в разделе 6 протокола, и согласованы с инструментом выравнивания множественных последовательностей Clustal Omega. Свойства аминокислот обозначаются цветом (красный, малый/гидрофобный; синий, кислый; пурпурный, основной; зеленый, гидроксил/сульфгидрил/амин) (далее определяется в таблице 2). Символы под аминокислотами указывают на степень сохранности (звездочки, полностью сохраненные остатки; толстые кишки, аминокислоты с сильно похожими свойствами; периоды сохранения между группами слабо сходных свойств) (подробно описано в таблице 1). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
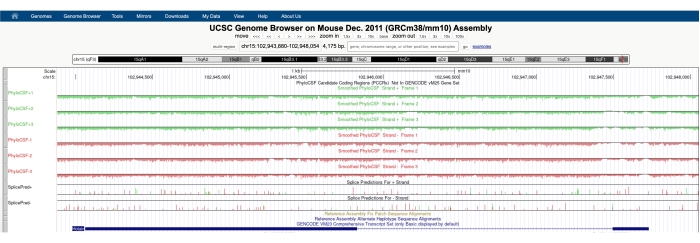
Рисунок 3: Скриншот треков PhyloCSF для проверенной длинной некодирующей РНК Hotair показывает отсутствие сохранения последовательности во всем ее геномном локусе. Наконечники стрел в интронной области Hotair направлены влево, указывая на то, что lncRNA транскрибируется из отрицательной цепи ДНК, и поэтому треки PhyloCSF -1, -2 и -3 должны быть в центре анализа. Обратите внимание, что оценка PhyloCSF отрицательна по всему гену (для всех шести треков), что указывает на отсутствие сохранения последовательности, что поддерживает его правильную аннотацию как некодирующей РНК. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 4: PhyloCSF анализ гена мыши 1810058I24Rik , который кодирует микропротеин mitolamban/Stmp1/Mm47. (A) Ген мыши 1810058I24Rik состоит из трех экзонов, а наконечники стрел в интронных областях указывают на то, что он транскрибируется на плюсовую нить ДНК и, следовательно, следует проанализировать треки PhyloCSF +1, +2 и +3. Законсервированная микропротеиновая кодирующая последовательность охватывает все три экзона, начиная с экзона 1 (B), считывая через экзон 2 (C) и заканчивая экзоном 3 (D). Обратите внимание, что положительная оценка PhyloCSF находится на треке +2 в экзоне 1, треке +3 в экзоне 2 и треке +2 в экзоне 1. Причина перемещения положительной оценки от одного трека к другому заключается в том, что PhyloCSF анализирует шесть потенциальных кадров чтения последовательности ДНК независимо от структуры экзона / интрона гена. Таким образом, интрон, содержащий ряд нуклеотидов, который не делится на три (три нуклеотида / кодон), вызовет сдвиг в кадре считывания в другую сторону. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 5: Анализ геномного локуса Mief1 с помощью PhyloCSF идентифицирует область с белково-кодирующим потенциалом в 5' UTR, которая не зависит от основного CDS Mief1 на общей РНК. Было показано, что этот сохраненный восходящий ORF (uORF) кодирует микропротеин под названием Mief1-MP. (A) Обзор геномного локуса Mief1 . Наконечники стрелок в интронах указывают направо, указывая на то, что Mief1 транскрибируется из плюсовой нити ДНК (сосредоточьтесь на треках PhyloCSF +1, +2 и +3 для определения потенциала кодирования). Основной CDS Mief1 кодирует 463 аминокислотных белка и показан на панели (B). Тем не менее, существует также отчетливый сохраненный восходящий ORF в 5'UTR Mief1, который кодирует уникальный микропротеин 70 аминокислот под названием Mief1-MP (C). Как видно из панели C, Mief1-MP имеет свой собственный сохраненный кодон старта и остановки в Mief1 5' UTR, а ORF очень высоко оценивается на треке PhyloCSF +1, что является убедительным доказательством того, что он кодирует функциональный микропротеин. Сокращения: ORF = открытая рамка чтения; uORF = восходящий ORF; UTR = непереведенный регион; CDS = последовательность кодирования. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
| Символ | Уровень сохранения аминокислот | Сгруппированные аминокислоты |
| Звездочка (*) | Полностью консервированный остаток | Не применимо (одиночный, полностью консервированный остаток) |
| Толстая кишка (:) | Группы со сильно похожими свойствами | СТА; NEQK; НХКК; НДЭК; КХРК; МИЛВ; ИФОМ; HY; FYW |
| Период (.) | Группы со слабо похожими свойствами | ККА; Квадроциклы; САГ; СТНК; СТПА; СГНД; СНДЭКК; НДЕКХК; НЕХРК; ФВЛИМ; HFY |
| Пробел (без символа) | Нет сходства | Не применимо (нет сходства) |
Таблица 1: Определения консенсусных символов для множественных выравниваний последовательностей, генерируемых Clustal Omega. Выравнивание последовательностей нескольких видов, показанное на рисунке 2 , было получено с использованием Clustal Omega52. Сокращения: серин (S), треонин (T), аланин (A), аспарагин (N), глутаминовая кислота (E), глутамин (Q), лизин (K), аспарагиновая кислота (D), аргинин (R), метионин (M), изолейцин (I), лейцин (L), фенилаланин (F), гистидин (H), тирозин (Y), триптофан (W), цистеин (C), валин (V), глицин (G), пролин (P).
| Цвет шрифта | Свойство | Аминокислотный остаток [Аббревиатура] |
| Красный | Малый, гидрофобный | аланин [A], валин [V], фенилаланин [F], пролин [P], метионин [M], изолейцин [I], лейцин [L], триптофан [W] |
| Синий | Кислотный | аспарагиновая кислота [D], глутаминовая кислота [E] |
| Пурпурный | Основной | аргинин [R], лизин [K] |
| Зеленый | Гидроксил, сульфгидрил, амин, +G | серин [S], треонин [T], тирозин [Y], гистидин [H], цистеин [C], аспарагин [N], глицин [G], глютамин [Q] |
Таблица 2: Свойства аминокислот, изображенных на рисунке 2. Clustal Omega52 использовался для генерации множественного выравнивания последовательностей, показанного на рисунке 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Протокол, представленный здесь, предоставляет подробные инструкции о том, как опрашивать геномные области, представляющие интерес для потенциала кодирования микропротеинами, с помощью PhyloCSF на удобном для пользователя UCSC Genome Browser 48,49,50,51. Как подробно описано выше, PhyloCSF является мощным алгоритмом сравнительной геномики, который объединяет филогенетические модели и частоты замещения кодонов для выявления эволюционных сигнатур, типичных для генов, кодирующих белки48,49. PhyloCSF широко используется для идентификации функциональных микропротеинов в геномных областях, ранее аннотированных как некодирующие 38,39,40,41,42,43,44,45,46,47 , и было показано, что этот подход превосходит другие методы сравнительной геномики для коротких последовательностей, таких как микропротеины размером до 13 аминокислот, и для небольших экзонов канонических белков 35,48,49. Примечательно, что полезность PhyloCSF как надежного метода идентификации функциональных белково-кодирующих последовательностей посредством эволюционного сохранения выходит за рамки позвоночных и беспозвоночных видов и даже недавно была применена к вирусным геномам для успешного опроса белковой кодирующей способности генома SARS-CoV-267.
В дополнение к идентификации предполагаемых кодирующих последовательностей в аннотированных некодирующих РНК, преимущество PhyloCSF заключается в том, что он также может надежно обнаруживать сохраненные микропротеины, кодируемые ORF в аннотированных нетранслируемых областях (UTR) канонических генов, кодирующих белки, включая как 5' вверх, так и 3' вниз по течению ORF (uORFs и dORFs, соответственно)8,19,66,68 . Например, микропротеин MIEF1 (MIEF1-MP) кодируется в 5' UTR фактора удлинения митохондрий 1 (MIEF1)66. В случае MIEF1-MP дискретный положительный балл PhyloCSF, соответствующий MIEF1-MP, наблюдается перед ORF, кодирующим MIEF1 (рисунок 5). В то время как некоторые микропротеины, кодируемые uORF, непосредственно взаимодействуют с нижестоящими каноническими белками на их общей мРНК (например, MIEF1-MP и MIEF1), другие функционируют независимо от белка, кодируемого основным CDS66,68. Поэтому при характеристике микропротеинов, кодируемых uORF, не следует предполагать, что они функционируют посредством прямой регуляции их последующего белкового продукта.
Хотя PhyloCSF имеет много явных сильных сторон в качестве инструмента для идентификации сохраненных микропротеин-кодирующих последовательностей, важно признать несколько ограничений этого метода. Во-первых, в то время как сохранение последовательности убедительно свидетельствует о том, что геномная область подверглась функциональному отбору и, таким образом, кодирует, отсутствие надежного сохранения и результирующая отрицательная оценка PhyloCSF не исключают окончательно кодирующего потенциала для данной последовательности. Другими словами, опора исключительно на PhyloCSF может привести к надзору за переведенными ORF, которые не сильно сохраняются, но все же производят функциональные микропротеины. Примечательно, что геномные области с низкими показателями сохранения или отрицательными показателями сохранения могут соответствовать видоспецифичным кодирующим областям или областям эволюционных «молодых» генов через дивергенцию последовательностей или рождение гена de novo 46,69,70,71,72,73,74. Например, микропротеин ASAP, который кодируется тем, что ранее считалось некодирующей РНК человека LINC00467, не оценивается положительно PhyloCSF, потому что аминокислотная последовательность сохраняется только у высших млекопитающих75. Кроме того, недавние исследования выявили несколько специфических для человека микропротеинов, в том числе один, кодируемый интергенной lncRNA RP3-527G5.1, которая не генерирует положительный балл PhyloCSF68,72. В связи с этим отсутствие положительной оценки PhyloCSF не может быть истолковано как доказательство некодирующей области и должно интерпретироваться с осторожностью.
Второе соображение, которое следует иметь в виду при использовании PhyloCSF, заключается в том, что, хотя положительная оценка в значительной степени наводит на мысль о функциональном отборе и способности кодировать белок, эта линия доказательств не может стоять отдельно и должна быть экспериментально подтверждена. Примеры методов, которые могут быть использованы для получения подтверждающих доказательств стабильной экспрессии микропротеинов, включают обнаружение предполагаемого белка с помощью масс-спектрометрии или западного блоттинга с использованием антитела, поднятого против интересующей микропротеиновой последовательности. В качестве альтернативы, поскольку может быть сложно генерировать надежные антитела для микропротеинов из-за отсутствия выбора последовательности для оптимальной антигенности, также можно использовать CRISPR / Cas9 и путь гомологической репарации (HDR) для введения эпитопной метки в эндогенный локус в рамках предполагаемой последовательности микропротеинов, тем самым облегчая обнаружение интересующего белка с использованием высокоаффинного антитела (например, ФЛАГ, HA, V5, Myc)18. Последнее ограничение PhyloCSF, которое следует признать, заключается в том, что, хотя в настоящее время он интегрирован во многие из широко используемых геномных сборок, включая Homo sapiens (человеческий hg19, hg38), Mus musculus (мышь mm10, mm39), Gallus gallus (курица, galGal4, galGal6), Drosophila melanogaster (плодовая муха, dm6), Caenorhabditis elegans (нематоды, ce11) и SARS-CoV-2 (wuhCor1) все еще есть много видов, которые в настоящее время не могут быть запрошены непосредственно в браузере генома UCSC.
Идентификация сохраненных доменов или характеристик последовательности в идентифицированных микропротеинах может помочь повысить уверенность в их функциональной значимости и дать некоторое представление об их предполагаемой функции. Здесь мы предоставляем рекомендации по конкретным инструментам и ресурсам, которые могут быть использованы для анализа идентифицированных последовательностей аминокислот микропротеинов более подробно, чтобы получить такое понимание. Конкретные инструменты, перечисленные ниже (и обобщенные в Таблице материалов), находятся в свободном доступе для общественности, и мы обнаружили, что они особенно удобны для пользователя и надежны в исследованиях микропротеинов 18,38,39,40,41,47. Помимо инструментов, описанных здесь, существует множество дополнительных ресурсов, которые можно найти на порталах ресурсов биоинформатики, таких как Expasy (https://www.expasy.org) и EMBL-EBI (https://www.ebi.ac.uk/services/all). Однако подробное описание специфики каждого из инструментов в этих репозиториях выходит за рамки данной статьи. Здесь мы рекомендуем следующие ресурсы.
Во-первых, TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) анализирует белковые последовательности, представляющие интерес, на наличие трансмембранных доменов. Примечательно, что ряд микропротеинов, которые были функционально охарактеризованы до сих пор, содержат однопроходные трансмембранные домены, что облегчает их локализацию в мембранных областях и позволяет их прямую регуляцию ионных каналов, обменников и мембраноассоциированных ферментов30. Во-вторых, Национальный центр биотехнологической информации (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) является популярным инструментом, используемым для идентификации сохраненных доменов в белковых или кодирующих нуклеотидных последовательностях. В-третьих, база данных семейства белков (Pfam)78 (http://pfam.xfam.org) предоставляет выравнивания и классификации семейств и доменов белков. В-четвертых, WoLF PSORT79 (https://wolfpsort.hgc.jp/) является инструментом, который может быть использован для прогнозирования субклеточной локализации белка. В-пятых, COXPRESdB80 представляет собой базу данных коэкспрессии генов (https://coxpresdb.jp), которая обеспечивает корегулируемые отношения генов для оценки функций генов. Наконец, SignalP 6.081 является широко используемой программой прогнозирования (https://services.healthtech.dtu.dk/service.php?SignalP), которая распознает наличие последовательности сигнальных пептидов и предсказывает местоположение места расщепления.
Таким образом, методы, описанные здесь, могут быть использованы для эффективного анализа геномных областей, представляющих интерес для белково-кодирующего потенциала, с использованием PhyloCSF в браузере генома UCSC. Эти методы очень доступны и могут быть легко изучены и эффективно применены людьми без предварительной подготовки или опыта в области биоинформатики или сравнительной геномики. Как подробно показано здесь, PhyloCSF является мощным инструментом, который может быть применен в качестве анализа первого прохода, чтобы помочь отличить кодирующие белок и некодирующие гены в геномах позвоночных, беспозвоночных и вирусных, и сильные стороны этого подхода значительно перевешивают отмеченные недостатки.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Авторы заявляют, что у них нет конкурирующих финансовых интересов.
Acknowledgments
Эта работа была поддержана грантами Национальных институтов здравоохранения (HL-141630 и HL-160569) и Фонда детских исследований Цинциннати (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
