

Summary
Het protocol dat hier wordt beschreven, biedt gedetailleerde instructies voor het analyseren van genomische gebieden die van belang zijn voor microproteïnecoderingspotentieel met behulp van PhyloCSF op de gebruiksvriendelijke UCSC Genome Browser. Daarnaast worden verschillende hulpmiddelen en middelen aanbevolen om de sequentiekenmerken van geïdentificeerde microproteïnen verder te onderzoeken om inzicht te krijgen in hun vermeende functies.
Abstract
Next-generation sequencing (NGS) heeft het veld van genomica vooruitgestuwd en hele genoomsequenties geproduceerd voor tal van diersoorten en modelorganismen. Ondanks deze schat aan sequentie-informatie zijn uitgebreide genannotatie-inspanningen echter een uitdaging gebleken, vooral voor kleine eiwitten. Met name conventionele eiwitannotatiemethoden werden ontworpen om opzettelijk vermeende eiwitten uit te sluiten die zijn gecodeerd door korte open leesframes (sORF's) met een lengte van minder dan 300 nucleotiden om het exponentieel hogere aantal onechte niet-coderende sORF's in het hele genoom te filteren. Als gevolg hiervan zijn honderden functionele kleine eiwitten genaamd microproteïnen (<100 aminozuren in lengte) ten onrechte geclassificeerd als niet-coderende RNA's of volledig over het hoofd gezien.
Hier bieden we een gedetailleerd protocol om gratis, openbaar beschikbare bio-informaticatools te gebruiken om genomische regio's te doorzoeken op microproteïne-codeerpotentieel op basis van evolutionair behoud. In het bijzonder bieden we stapsgewijze instructies over het onderzoeken van sequentiebehoud en coderingspotentieel met behulp van fylogenetische codonsubstitutiefrequenties (PhyloCSF) op de gebruiksvriendelijke Genome Browser van de University of California Santa Cruz (UCSC). Daarnaast beschrijven we stappen om efficiënt meerdere soorten uitlijningen van geïdentificeerde microproteïnesequenties te genereren om aminozuursequentiebehoud te visualiseren en middelen aan te bevelen om microproteïnekenmerken te analyseren, inclusief voorspelde domeinstructuren. Deze krachtige hulpmiddelen kunnen worden gebruikt om vermeende microproteïne-coderende sequenties in niet-canonische genomische regio's te helpen identificeren of om de aanwezigheid van een geconserveerde coderingssequentie met translationeel potentieel in een niet-coderend transcript van belang uit te sluiten.
Introduction
De identificatie van de volledige set coderende elementen in het genoom is een belangrijk doel sinds de start van het Human Genome Project en blijft een centrale doelstelling voor het begrijpen van biologische systemen en de etiologie van genetisch gebaseerde ziekten 1,2,3,4. Vooruitgang in NGS-technieken heeft geleid tot de productie van hele genoomsequenties voor een groot aantal organismen, waaronder gewervelde dieren, ongewervelde dieren, gist en planten5. Bovendien hebben transcriptionele sequencingmethoden met hoge doorvoer de complexiteit van het cellulaire transcriptoom verder onthuld en duizenden nieuwe RNA-moleculen geïdentificeerd met zowel eiwitcoderende als niet-coderende functies 6,7. Het decoderen van deze enorme hoeveelheid sequentie-informatie is een continu proces en er blijven uitdagingen bestaan met uitgebreide genannotatie-inspanningen8.
De recente ontwikkeling van translationele profileringsmethoden, waaronder ribosoomprofilering 9,10 en poly-ribosoomsequencing11, hebben bewijs geleverd waaruit blijkt dat honderden niet-canonische translatiegebeurtenissen worden toegewezen aan momenteel niet-geannoteerde sORF's in het hele genoom, met het potentieel om kleine eiwitten te genereren die microproteïnen of micropeptiden worden genoemd 12,13,14,15,16, 17. Microproteïnen zijn naar voren gekomen als een nieuwe klasse van veelzijdige eiwitten die eerder over het hoofd werden gezien door standaard genannotatiemethoden vanwege hun kleine omvang (<100 aminozuren) en het ontbreken van klassieke eiwitcoderende genkenmerken 8,12,18,19,20. Microproteïnen zijn beschreven in vrijwel alle organismen, waaronder gist21,22, vliegen 17,23,24 en zoogdieren 25,26,27,28, en er is aangetoond dat ze een cruciale rol spelen in diverse processen, waaronder ontwikkeling, metabolisme en stress signalering 19,20,29, 30,31,32,33,34. Het is dus noodzakelijk om het genoom te blijven ontginnen voor extra leden van deze lang over het hoofd geziene klasse van functionele kleine eiwitten.
Ondanks de wijdverspreide erkenning van het biologische belang van microproteïnen, blijft deze klasse van genen enorm ondervertegenwoordigd in genoomannotaties, en hun nauwkeurige identificatie blijft een voortdurende uitdaging die de vooruitgang in het veld heeft belemmerd. Verschillende computationele hulpmiddelen en experimentele methoden zijn onlangs ontwikkeld om de moeilijkheden te overwinnen die gepaard gaan met het identificeren van microproteïne-coderende sequenties (uitgebreid besproken in verschillende uitgebreide beoordelingen 8,35,36,37). Veel recente microproteïne-identificatiestudies 38,39,40,41,42,43,44,45,46,47 hebben sterk vertrouwd op het gebruik van een dergelijk algoritme genaamd PhyloCSF48,49 , een krachtige vergelijkende genomicabenadering die kan worden gebruikt om geconserveerde eiwitcoderende regio's van het genoom te onderscheiden van die welke niet-coderend zijn.
PhyloCSF vergelijkt codonsubstitutiefrequenties (CSF) met behulp van multi-species nucleotide-uitlijningen en fylogenetische modellen om evolutionaire handtekeningen van eiwitcoderende genen te detecteren. Deze empirische modelgebaseerde benadering is gebaseerd op het uitgangspunt dat eiwitten voornamelijk worden geconserveerd op aminozuurniveau in plaats van de nucleotidesequentie. Daarom worden synonieme codonsubstituties, die hetzelfde aminozuur coderen, of codonsubstituties naar aminozuren met geconserveerde eigenschappen (d.w.z. lading, hydrofobiciteit, polariteit) positief beoordeeld, terwijl niet-synonieme substituties, inclusief missense en onzinsubstituties, negatief scoren. PhyloCSF is getraind op gegevens over het hele genoom en heeft bewezen effectief te zijn in het scoren van korte delen van een coderende sequentie (CDS) in isolatie van de volledige sequentie, wat nodig is bij het analyseren van microproteïnen of individuele exonen van standaard eiwitcoderende genen48,49.
Met name de recente integratie van de PhyloCSF-trackhubs in de Genome Browser 49,50,51 van de University of California Santa Cruz (UCSC) stelt onderzoekers van alle achtergronden in staat om eenvoudig toegang te krijgen tot een gebruiksvriendelijke interface om genomische regio's te doorzoeken die van belang zijn voor eiwitcoderingspotentieel. Het onderstaande protocol biedt gedetailleerde instructies over het laden van de PhyloCSF-trackhubs op de UCSC Genome Browser en vervolgens genomische regio's van belang te onderzoeken om te onderzoeken op zeer betrouwbare eiwitcoderende regio's (of het gebrek daaraan). Bovendien, in het geval dat een positieve PhyloCSF-score wordt waargenomen, worden stappen afgebakend om het microproteïne-coderende potentieel verder te analyseren en efficiënt meerdere soorten uitlijningen van de geïdentificeerde aminozuursequenties te genereren om het behoud van de sequenties tussen soorten te illustreren. Ten slotte worden verschillende aanvullende openbaar beschikbare middelen en hulpmiddelen geïntroduceerd in de discussie om geïdentificeerde microproteïnekenmerken te onderzoeken, waaronder voorspelde domeinstructuren en inzicht in vermeende microproteïnefunctie.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Het protocol dat hieronder wordt beschreven, beschrijft stappen voor het laden en navigeren van de PhyloCSF-browsertracks op de UCSC Genome Browser (gegenereerd door Mudge et al.49). Voor algemene vragen over de UCSC Genome Browser is hier een uitgebreide Genome Browser User's Guide te vinden: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. De PhyloCSF Track Hub laden in de UCSC Genome Browser
- Open een internetbrowservenster en navigeer naar de UCSC Genome Browser (https://genome.ucsc.edu/).
- Selecteer onder de kop Onze tools de optie Hubs volgen .
OPMERKING: De optie Hubs volgen is ook te vinden op het tabblad Mijn gegevens . - Typ op het tabblad Public Hubs PhyloCSF in het vak Zoektermen . Klik op de knop Openbare hubs zoeken .
- Maak verbinding met PhyloCSF door op de knop Verbinden te klikken voor de hubnaam PhyloCSF (Beschrijving: Evolutionair eiwitcoderingspotentieel zoals gemeten door PhyloCSF).
OPMERKING: Deze Track Hub wordt geladen voor tal van assemblages, waaronder mensen (hg19 en hg38) en muizen (mm10 en mm39). - Nadat u op verbinden hebt geklikt, wacht u om te worden omgeleid naar de UCSC Genome Browser Gateway-pagina (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Navigeren naar interessante genen met behulp van gene identifiers
- Selecteer de soort en het genoomassemblage om op te vragen. Als u een andere soort wilt opvragen (bijvoorbeeld de muis), selecteert u de gewenste soort onder de kop Bladeren/Soort selecteren door op het juiste pictogram te klikken, of typt u de soort in het tekstvak met de tekst : Voer soort, algemene naam of verzamel-ID in.
OPMERKING: De assembly wordt direct weergegeven onder de kop Positie zoeken . Meestal is de standaard de Human Assembly (bijv. december 2009 [GRCh37/hg19]). - Kies de assembly die u wilt zoeken onder de kop Positie zoeken met behulp van het vervolgkeuzemenu.
- Voer de positie, het gensymbool of de zoekterm in het vak Positie/Zoekterm in en klik op Ga om naar een interessant gen in de Genome Browser te navigeren.
- Als de zoekopdracht meerdere overeenkomsten heeft opgeleverd, wacht u om te worden omgeleid naar een pagina waarvoor een interessante positie moet worden geselecteerd. Klik op het juiste gen van belang.
3. Navigeren naar genomische gebieden van belang met behulp van sequentie-informatie
- Navigeer naar de UCSC Genome Browser (https://genome.ucsc.edu/) en selecteer de BLAST-Like Alignment Tool (BLAT) onder de kop Onze tools om een specifieke DNA- of eiwitsequentie op te vragen. U kunt ook de cursor op het tabblad Extra plaatsen en de optie Blat selecteren of deze link volgen: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Selecteer de soort (Genoom) en De samenstellen van belang met behulp van de vervolgkeuzemenu's.
- Definieer het querytype met behulp van het vervolgkeuzemenu.
- Plak de volgorde van interesse in het tekstvak BLAT Search Genome en klik op Verzenden.
- Klik op de browserlink onder de kop ACTIES om naar de genomische regio van interesse te navigeren.
4. Het identificeren van geconserveerde sORFs met behulp van PhyloCSF Track Data
- Scan visueel het genomische interessegebied op positief scorende PhyloCSF-regio's (figuur 1).
OPMERKING: Voor een gedetailleerde uitleg van het visueel interpreteren van PhyloCSF-scores op de UCSC Genome Browser, zie de sectie representatieve resultaten hieronder. - Gebruik de zoomfunctie om interessante gebieden te vergroten om sequentiekenmerken te onderzoeken en te zoeken naar start/stop-codons. Als u handmatig wilt inzoomen, houdt u de Shift-toets ingedrukt en houdt u de muisknop ingedrukt terwijl u langs het interessegebied sleept. U kunt ook de inzoom- en uitzoomknoppen boven aan de pagina gebruiken om te navigeren (er zijn opties voor 1,5x, 3x, 10x of basiszoom beschikbaar).
OPMERKING: Voordat u de inzoom- / uitzoomknoppen gebruikt, is het noodzakelijk om het gen te verplaatsen zodat het interessegebied zich in het midden van het scherm bevindt. Om deze actie uit te voeren, klikt u op de afbeelding en sleept u deze naar links of rechts om het genomische gebied horizontaal naar wens te verplaatsen of gebruikt u de bewegingspijlen boven aan de pagina. - Zoom in totdat de nucleotide (basis) sequentie zichtbaar is.
OPMERKING: De nucleotidesequentie verschijnt direct boven de +1 Smoothed PhyloCSF-score. - Scan visueel de nucleotidesequentie aan het begin en einde van de positief scorende PhyloCSF-regio's om vermeende start (ATG) en stop (TGA / TAA / TAG) codons te identificeren.
OPMERKING: Als het gen van belang zich op de minus-streng van DNA bevindt, zijn de start- en stopcodons het omgekeerde complement (d.w.z. CAT voor het startcodon en TCA / TTA / CTA voor het stopcodon).
5. Homologe regio's bekijken in andere genomen
- Beweeg de muis over de kop Weergave boven aan de pagina en klik op de optie In Andere genomen (Converteren).
- Definieer het genoom van belang met behulp van het vervolgkeuzemenu onder de kop Nieuw genoom .
- Selecteer de genomische verzameling van belang met behulp van het vervolgkeuzemenu onder de kop Nieuwe assembly en klik vervolgens op de knop Verzenden .
- Zodra de browser een lijst met regio's in de nieuwe assemblage met gelijkenis retourneert, klikt u op de link naar de chromosoompositie om naar het homologe gebied van interesse te navigeren.
OPMERKING: Het percentage van de totale basen (nucleotiden) en de spanwijdte die door de regio worden bestreken, worden gedefinieerd voor elke vermelde regio. Hoe hoger het percentage matchingbases, hoe hoger de conservering is voor de regio van belang. - Volg dezelfde navigatiestrategieën die in sectie 4 worden beschreven om de volgorde te analyseren.
6. Het genereren van multi-species sequence alignments voor microproteïnen van belang
- Klik op het gen van belang in de GENCODE-track in de UCSC Genome Browser (aangegeven in figuur 1A met een blauw vak) om naar de genbeschrijvingspagina te navigeren.
- Klik onder de kop Volgorde en koppelingen naar gereedschappen en databases op de link in de tabel met de tekst Andere soorten FASTA.
- Klik op de vakjes die zijn gekoppeld aan de soort van belang om ze te selecteren. Klik op Verzenden. Kopieer en plak de reeksen die onder aan de pagina in FASTA-indeling worden weergegeven in een tekstverwerkingsdocument.
- Open een tweede browservenster en navigeer naar de Clustal Omega Multiple Sequence Alignment tool52 op de website van het European Bioinformatics Institute (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Plak de reeksbestanden die zich nog op het klembord bevinden in het vak in stap 1 waarin reeksen in een ondersteunde indeling worden gelezen. Scrol naar de onderkant van de pagina en klik op Verzenden. Kijk hieronder de uitgelijnde resultaten (in zwart lettertype) voor symbolen die de mate van conservering van elk aminozuur aangeven (symbolen zijn gedefinieerd in tabel 1).
OPMERKING: Het kan enkele minuten duren om de uitlijning te genereren. - Om de aminozuureigenschappen in kleur te bekijken, klikt u op de link Kleuren weergeven direct boven de sequenties om de aminozuren te kleuren volgens hun eigenschappen (gedefinieerd in tabel 2).
- Kopieer en plak de volgorde-uitlijning in een tekstverwerkings- of diavoorstellingsprogramma om een figuur of illustratiebestand te genereren (bijvoorbeeld figuur 2).
OPMERKING: Gebruik een monospaced lettertype voor de uitlijning, zoals Courier. - Om andere resultaten van de Clustal Omega-resultatenpagina te bekijken, klikt u op de juiste tabbladen (d.w.z. Guide Tree of Phylogenetic Tree).
- Klik op het tabblad Resultatenviewers voor opties om de reeksinformatie te bekijken met Jalview, een gratis programma dat gespecialiseerd is in het bewerken, visualiseren en analyseren van meerdere sequenties55, of om toegang te krijgen tot directe links naar MView en Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Hier zullen we het gevalideerde microproteïne mitoregulin (Mtln) als voorbeeld gebruiken om aan te tonen hoe een geconserveerde sORF een positieve PhyloCSF-score zal genereren die gemakkelijk kan worden gevisualiseerd en geanalyseerd op de UCSC Genome Browser. Mitoregulin werd eerder geannoteerd als een niet-coderend RNA (voorheen menselijk gen ID LINC00116 en muisgen ID 1500011K16Rik). Vergelijkende genomica en sequentieconserveringsanalysemethoden speelden een cruciale rol bij de eerste ontdekking 40,57,58,59,60,61, wat de kracht van deze methoden benadrukt. Voor dit voorbeeld wordt de muis GRCm38/mm10 (december 2011) assembly gebruikt. De zoekopdracht kan worden uitgevoerd met behulp van de gen-id's (mitoregulin, Mtln) of de genpositie (chr2:127,791,364-127,792,496) zoals beschreven in protocol sectie 2. Als alternatief kan de aminozuursequentie voor mitoreguline (weergegeven in figuur 2) worden doorzocht met behulp van de BLAT-tool (beschreven in protocolsectie 3).
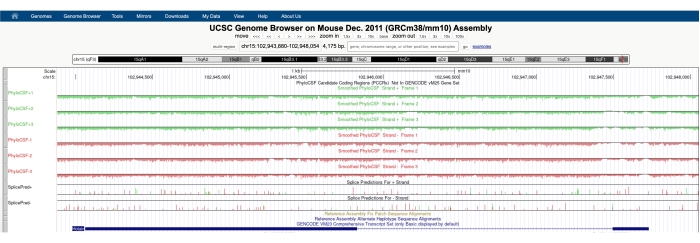
Een scherm dat lijkt op het scherm in figuur 1A wordt weergegeven met de PhyloCSF Track Hub zichtbaar bovenaan het scherm. De Smoothed PhyloCSF-tracks (gladgestreken met een verborgen Markov-model dat een waarschijnlijkheid definieert dat elk codon codeert) worden afgebeeld als zes totale tracks, met drie sporen die overeenkomen met de plusstreng van DNA (in het groen afgebeeld als PhyloCSF +1, +2 en +3) en drie sporen die overeenkomen met de minusstreng van DNA (in rood afgebeeld als PhyloCSF -1, -2 en -3). Deze tracks vertegenwoordigen de drie potentiële leesframes voor het gen van belang in elke richting. In het browservenster worden exonen afgebeeld als blauwe rechthoeken verbonden door dunne blauwe horizontale lijnen, die de introns vertegenwoordigen. De pijlpunten op de intronische gebieden geven aan in welke richting het gen wordt getranscribeerd (en dus op welke streng moet worden gefocust voor de PhyloCSF-score). Voor het voorbeeld van Mtln in figuur 1 wijzen de intronische pijlpunten naar links. Daarom wordt het Mtln-gen getranscribeerd uit de minus-streng van DNA en wordt de relevante PhyloCSF-score weergegeven in de -1, -2 en -3 sporen (in rood).
Elke PhyloCSF-track wordt weergegeven als een dunne zwarte lijn met negatieve scoregebieden weergegeven in lichtgroen / rood onder de lijn en positieve scoregebieden aangegeven in donkergroen / rood boven de lijn. Zoals beschreven in de inleiding, geeft een positieve PhyloCSF-score een geconserveerd gebied aan dat waarschijnlijk codeert. Merk op dat voor eiwitcoderende regio's met een bijzonder hoge sequentieconservering, ze vaak ook positief scoren op de antisense-streng; de PhyloCSF-score is echter meestal hoger op de juiste streng. Dit is bijvoorbeeld te zien in figuur 1 voor Mtln, waar de juiste coderingsvolgorde zeer hoog scoort in de PhyloCSF -1-track en de antisense-streng (PhyloCSF +2-track) ook een positieve score genereert. Zoals te zien is in figuur 1A (aangegeven met zwarte doos), is er een gebied in het eerste exon van Mtln dat zeer hoog scoort op de PhyloCSF -1-track, wat suggereert dat dit kan overeenkomen met een coderingsgebied. Om dit gebied nader te onderzoeken, is het handig om in te zoomen en het gebied te vergroten (figuur 1B). Zoals te zien is in figuur 1C,D, begint het positief scorende gebied in het eerste exon van Mtln direct over een startcodon (figuur 1C) en eindigt het bij een stopcodon (figuur 1D), wat aangeeft dat deze ORF sterk geconserveerd is en sterk suggereert dat het een coderende ORF is. Aangezien Mtln zich op de min-streng van DNA bevindt, worden de start- en stopcodons weergegeven als het omgekeerde complement van het codon (d.w.z. het ATG-startcodon wordt weergegeven als CAT [figuur 1C] en het TGA-stopcodon wordt weergegeven als TCA [figuur 1D]).
Naast het gebruik van PhyloCSF om te zoeken naar geconserveerde gebieden met microproteïnecoderingspotentieel, kan deze techniek ook worden toegepast als een first-pass analyse van vermeende niet-coderende RNA's om de aanwezigheid van een geconserveerde ORF uit te sluiten, waardoor ondersteuning wordt geboden voor een niet-coderende annotatie. Analyse van het goed gekarakteriseerde lncRNA HOTAIR 62,63 met behulp van PhyloCSF toont bijvoorbeeld een negatieve score in het hele gen over alle zes sporen (figuur 3), wat sterk wijst op een gebrek aan sequentiebehoud en ondersteuning biedt dat HOTAIR correct is geannoteerd als een niet-coderend RNA.
Zoals duidelijk te zien is in figuur 1, bevindt de volledige coderende ORF voor mitoregulin zich binnen één exon, waardoor een eenvoudige en duidelijke uitlezing door PhyloCSF wordt geproduceerd met een enkele, ononderbroken, positief scorende regio. PhyloCSF-trackhubgegevens zijn echter niet altijd even duidelijk en gemakkelijk te interpreteren. Het mitolamban/Stmp1/Mm47 microproteïne gecodeerd door de muis 1810058I24Rik gen 47,64,65 toont bijvoorbeeld een geconserveerde ORF die drie exonen omvat (Figuur 4A), en de positieve PhyloCSF score springt van de +2 track in exon 1 (Figuur 4B) naar de +3 track in exon 2 (Figuur 4C), en dan terug naar de +2 track in exon 3 (Figuur 4D ). Hoewel dit op het eerste gezicht verwarrend lijkt, is de verklaring vrij eenvoudig. PhyloCSF scoort de zes potentiële leesframes (drie op de plusstreng van DNA en drie op de minstreng) van genomische regio's zonder rekening te houden met de specifieke exon / intron-architectuur voor elk gen. Daarom behoudt het de intronische sequentie-informatie in de 3-nucleotide periodiciteit van de leesframes. Dus als een intron een aantal nucleotiden bevat die niet deelbaar zijn door drie (d.w.z. drie nucleotiden / codon), zal het PhyloCSF-leesframe van het ene spoor naar het andere springen.
Ten slotte kan PhyloCSF ook effectief worden gebruikt om meerdere verschillende coderende ORF's binnen een enkel RNA-molecuul te identificeren. Het MIEF1-microproteïne (MIEF1-MP) is bijvoorbeeld gecodeerd binnen de 5' UTR van mitochondriale rekfactor 1 (MIEF1)66 (figuur 5). Wanneer het MIEF1-genomische gebied wordt geanalyseerd door PhyloCSF, kan een discrete positieve PhyloCSF-score die overeenkomt met de MIEF1-MP (figuur 5C) gemakkelijk worden waargenomen vóór de hoofd-CDS voor MIEF1 (figuur 5B). Verdere discussie over MIEF1 en het bijbehorende microproteïne (MIEF1-MP) wordt hieronder in de discussie gegeven, samen met een samenvatting van de sterke en zwakke punten van de methoden en protocollen die in dit artikel worden beschreven.

Figuur 1: FylokoCSF-analyse van het mitoreguline (Mtln)-gen geeft een gebied aan met een hoge sequentieconservatie dat overeenkomt met een gevalideerd microproteïne. (A) Screenshots van de UCSC Genome Browser en PhyloCSF Tracks laten zien dat Mtln twee exonen en een enkele intron bevat. De pijlpunten in het intron wijzen naar links, wat aangeeft dat het Mtln-gen is getranscribeerd uit de minus-streng van DNA, en de relevante PhyloCSF-scores worden daarom weergegeven in de -1, -2 en -3 tracks (in rood). De volledige mitoregulin-coderingssequentie bevindt zich in Exon 1 en scoort hoog op de PhyloCSF -1-track (B). Een geconserveerd startcodon kan duidelijk worden waargenomen aan het begin van het positief scorende gebied in de PhyloCSF -1-track (C), die is gemarkeerd met een groen vak (CAT, reverse complement ATG). Bovendien wordt een geconserveerd stopcodon (TCA, omgekeerd complement TGA) aangegeven met een rood vak in paneel (D), dat overeenkomt met het einde van het positief scorende PhyloCSF-gebied. Gedetailleerde informatie over het Mtln-gen is te vinden door te klikken op de Mtln-genidentificatie in het blauwe vak (weergegeven in paneel A). Opmerkelijk is dat sterk geconserveerde eiwitcoderende regio's vaak ook positief scoren op de antisense-streng (hier te zien in de PhyloCSF +2-track voor Mtln). De PhyloCSF-score is echter meestal hoger op de juiste streng (de PhyloCSF -1-track in dit voorbeeld). Klik hier om een grotere versie van deze figuur te bekijken.

Figuur 2: Meerdere soorten sequentie-uitlijning van het microproteïne mitoregulin gegenereerd met behulp van het Clustal Omega-programma. De mitoreguline aminozuursequenties voor de acht aangegeven soorten werden geëxtraheerd zoals beschreven in protocolsectie 6 en afgestemd op de Clustal Omega multiple sequence alignment tool. De eigenschappen van de aminozuren worden aangegeven door kleur (rood, klein/hydrofoob; blauw, zuur; magenta, basisch; groen, hydroxl/sulfhydryl/amine) (verder gedefinieerd in tabel 2). De symbolen onder de aminozuren geven de mate van conservering aan (sterretjes, volledig geconserveerde residuen; dubbele punten, aminozuren met sterk vergelijkbare eigenschappen; perioden, behoud tussen groepen met zwak vergelijkbare eigenschappen) (uitgebreid beschreven in tabel 1). Klik hier om een grotere versie van deze figuur te bekijken.
Figuur 3: Een screenshot van de PhyloCSF-tracks voor het gevalideerde lange niet-coderende RNA Hotair toont een gebrek aan sequentiebehoud gedurende de genomische locus. De pijlpunten in het intronische gebied van Hotair wijzen naar links, wat aangeeft dat het lncRNA is getranscribeerd uit de negatieve dna-streng, en daarom moeten de PhyloCSF -1, -2 en -3 sporen de focus van de analyse zijn. Merk op dat de PhyloCSF-score negatief is in het hele gen (voor alle zes tracks), wat wijst op een gebrek aan sequentiebehoud, wat de juiste annotatie als een niet-coderend RNA ondersteunt. Klik hier om een grotere versie van deze figuur te bekijken.

Figuur 4: FylokoCSF-analyse van het muis 1810058I24Rik-gen , dat codeert voor het microproteïne mitolamban / Stmp1 / Mm47. (A) Het muis 1810058I24Rik-gen bestaat uit drie exonen en de pijlpunten in de intronische gebieden wijzen naar rechts, wat aangeeft dat het is getranscribeerd op de plusstreng van DNA en daarom moeten de PhyloCSF +1, +2 en +3 sporen worden geanalyseerd. De geconserveerde microproteïnecoderingssequentie omvat alle drie de exonen, beginnend in exon 1 (B), lezend via exon 2 (C) en eindigend in exon 3 (D). Merk op dat de positieve PhyloCSF-score wordt gevonden op de +2-track in exon 1, de +3-track in exon 2 en de +2-track in exon 1. De reden voor de beweging van de positieve score van het ene spoor naar het andere is dat PhyloCSF de zes potentiële leesframes van de DNA-sequentie analyseert, onafhankelijk van de exon / intronstructuur van het gen. Daarom zal een intron met een aantal nucleotiden dat niet deelbaar is door drie (drie nucleotiden/codon) een verschuiving in het leesframe naar een ander spoor veroorzaken. Klik hier om een grotere versie van deze figuur te bekijken.

Figuur 5: Analyse van de Mief1 genomische locus met PhyloCSF identificeert een gebied met eiwitcoderend potentieel in de 5' UTR dat onafhankelijk is van het belangrijkste Mief1 CDS op het gedeelde RNA. Van deze geconserveerde upstream ORF (uORF) is aangetoond dat het een microproteïne codeert met de naam Mief1-MP. (A) Overzicht van de Mief1 genomische locus. De pijlpunten in de introns wijzen naar rechts, wat aangeeft dat Mief1 is getranscribeerd uit de plusstreng van DNA (focus op de PhyloCSF +1, +2 en +3 tracks om het coderingspotentieel te bepalen). De belangrijkste Mief1 CDS codeert voor een 463 aminozuureiwit en wordt weergegeven in paneel (B). Er is echter ook een duidelijke geconserveerde upstream ORF binnen de 5 'UTR van Mief1 die codeert voor een uniek 70 aminozuur microproteïne genaamd Mief1-MP (C). Zoals te zien is in Panel C, heeft de Mief1-MP zijn eigen geconserveerde start- en stopcodon binnen de Mief1 5 'UTR, en de ORF scoort zeer hoog op de PhyloCSF + 1-track, wat sterk bewijs levert dat het codeert voor een functioneel microproteïne. Afkortingen: ORF = open leeskader; uORF = stroomopwaarts ORF; UTR = onvertaald gebied; CDS = coderingsvolgorde. Klik hier om een grotere versie van deze figuur te bekijken.
| Symbool | Niveau van aminozuurbehoud | Gegroepeerde aminozuren |
| Sterretje (*) | Volledig geconserveerd residu | Niet van toepassing (enkelvoudig, volledig geconserveerd residu) |
| Dubbele punt (:) | Groepen met sterk vergelijkbare eigenschappen | Sta; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FYW |
| Punt (.) | Groepen met zwak vergelijkbare eigenschappen | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; Hfy |
| Spatie (geen symbool) | Geen gelijkenis | Niet van toepassing (geen gelijkenis) |
Tabel 1: Definities van consensussymbolen voor multiple sequence alignments gegenereerd door Clustal Omega. De uitlijning van meerdere soorten in figuur 2 werd gegenereerd met behulp van Clustal Omega52. Afkortingen: serine (S), threonine (T), alanine (A), asparagine (N), glutaminezuur (E), glutamine (Q), lysine (K), asparaginezuur (D), arginine (R), methionine (M), isoleucine (I), leucine (L), fenylalanine (F), histidine (H), tyrosine (Y), tryptofaan (W), cysteïne (C), valine (V), glycine (G), proline (P).
| Tekstkleur | Eigenschap | Amino Acid Residue [Afkorting] |
| Rood | Klein, hydrofoob | alanine [A], valine [V], fenylalanine [F], proline [P], methionine [M], isoleucine [I], leucine [L], tryptofaan [W] |
| Blauw | Zuur | asparaginezuur [D], glutaminezuur [E] |
| Magenta | Basisch | arginine [R], lysine [K] |
| Groen | Hydroxl, sulfhydryl, amine, +G | serine [S], threonine [T], tyrosine [Y], histidine [H], cysteïne [C], asparagine [N], glycine [G], glutamine [Q] |
Tabel 2: Eigenschappen van de aminozuren weergegeven in figuur 2. Clustal Omega52 werd gebruikt om de meervoudige sequentie-uitlijning in figuur 2 te genereren.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Het hier gepresenteerde protocol biedt gedetailleerde instructies over het ondervragen van genomische regio's die van belang zijn voor microproteïnecoderingspotentieel met behulp van PhyloCSF op de gebruiksvriendelijke UCSC Genome Browser 48,49,50,51. Zoals hierboven beschreven, is PhyloCSF een krachtig vergelijkend genomics-algoritme dat fylogenetische modellen en codonsubstitutiefrequenties integreert om evolutionaire handtekeningen te identificeren die typerend zijn voor eiwitcoderende genen48,49. PhyloCSF is op grote schaal gebruikt om functionele microproteïnen te identificeren in genomische regio's die eerder zijn geannoteerd als niet-coderend 38,39,40,41,42,43,44,45,46,47 , en van deze benadering is aangetoond dat deze beter presteert dan andere vergelijkende genomicamethoden voor korte sequenties zoals microproteïnen zo klein als 13 aminozuren en voor kleine exonen van canonieke eiwitten 35,48,49. Met name het nut van PhyloCSF als een robuuste methode om functionele eiwitcoderende sequenties te identificeren via evolutionair behoud gaat verder dan dat van gewervelde en ongewervelde soorten en is zelfs onlangs toegepast op virale genomen om met succes de eiwitcoderende capaciteit van het SARS-CoV-2-genoom te ondervragen67.
Naast het identificeren van vermeende coderende sequenties binnen geannoteerde niet-coderende RNA's, is een voordeel van PhyloCSF dat het ook op betrouwbare wijze geconserveerde microproteïnen kan detecteren die zijn gecodeerd door ORF's binnen geannoteerde niet-vertaalde regio's (UTR's) van canonieke eiwitcoderende genen, waaronder zowel 5'upstream als 3' downstream ORF's (respectievelijk uORF's en dORFs)8,19,66,68 . Het MIEF1-microproteïne (MIEF1-MP) is bijvoorbeeld gecodeerd in de 5' UTR van mitochondriale rekfactor 1 (MIEF1)66. In het geval van MIEF1-MP wordt een discrete positieve PhyloCSF-score waargenomen die overeenkomt met de MIEF1-MP vóór de ORF die MIEF1 codeert (figuur 5). Terwijl sommige uORF-gecodeerde microproteïnen rechtstreeks interageren met de downstream canonieke eiwitten op hun gedeelde mRNA (bijv. MIEF1-MP en MIEF1), functioneren andere onafhankelijk van het eiwit gecodeerd door de belangrijkste CDS66,68. Daarom moet bij het karakteriseren van uORF-gecodeerde microproteïnen niet worden aangenomen dat ze functioneren via directe regulatie van hun downstream eiwitproduct.
Hoewel PhyloCSF veel duidelijke sterke punten heeft als hulpmiddel voor de identificatie van geconserveerde microproteïne-coderende sequenties, is het belangrijk om verschillende beperkingen van deze methode te herkennen. Ten eerste, hoewel sequentiebehoud sterk suggereert dat een genomisch gebied functionele selectie heeft ondergaan en dus codeert, sluit een gebrek aan robuuste conservering en een resulterende negatieve PhyloCSF-score niet definitief uit dat het coderingspotentieel voor een bepaalde sequentie wordt uitgesloten. Met andere woorden, uitsluitend vertrouwen op PhyloCSF kan resulteren in het toezicht op vertaalde ORF's die niet sterk geconserveerd zijn, maar nog steeds functionele microproteïnen produceren. Met name genomische regio's met lage instandhoudings- of negatieve instandhoudingsscores kunnen overeenkomen met soortspecifieke coderende regio's of die van evolutionaire "jonge" genen via sequentie divergentie of de novo gengeboorte 46,69,70,71,72,73,74. Bijvoorbeeld, het microproteïne ASAP, dat wordt gecodeerd door wat vroeger werd beschouwd als het menselijke niet-coderende RNA LINC00467, wordt niet positief gescoord door PhyloCSF omdat de aminozuursequentie alleen wordt bewaard bij hogere zoogdieren75. Bovendien identificeerden recente studies verschillende mensspecifieke microproteïnen, waaronder een gecodeerd door het intergene lncRNA RP3-527G5.1, dat geen positieve PhyloCSF-score68,72 genereert. In dit opzicht kan de afwezigheid van een positieve PhyloCSF-score niet worden geïnterpreteerd als bewijs van een niet-coderend gebied en moet het met de nodige voorzichtigheid worden geïnterpreteerd.
Een tweede overweging om in gedachten te houden bij het gebruik van PhyloCSF is dat hoewel een positieve score zeer suggestief is voor functionele selectie en eiwitcoderende capaciteit, deze bewijslijn niet op zichzelf kan staan en experimenteel moet worden gevalideerd. Voorbeelden van methoden die kunnen worden gebruikt om ondersteunend bewijs te genereren voor stabiele microproteïne-expressie zijn de detectie van het vermeende eiwit door massaspectrometrie of western blotting met behulp van een antilichaam dat tegen de microproteïnesequentie van belang is verhoogd. Als alternatief, omdat het een uitdaging kan zijn om betrouwbare antilichamen voor microproteïnen te genereren vanwege het gebrek aan sequentiekeuzes voor optimale antigeniciteit, is het ook mogelijk om CRISPR / Cas9 en de homologie-gerichte reparatie (HDR) -route te gebruiken om een epitooptag in de endogene locus te introduceren in het kader van de vermeende microproteïnesequentie, waardoor de detectie van het eiwit van belang met behulp van een antilichaam met hoge affiniteit wordt vergemakkelijkt (bijv. VLAG, HA, V5, Myc)18. Een laatste beperking van PhyloCSF om te erkennen is dat hoewel het momenteel is geïntegreerd in veel van de veelgebruikte genomische assemblages, waaronder Homo sapiens (menselijk hg19, hg38), Mus musculus (muis mm10, mm39), Gallus gallus (kip, galGal4, galGal6), Drosophila melanogaster (fruitvlieg, dm6), Caenorhabditis elegans (nematoden, ce11) en SARS-CoV-2 (wuhCor1), er zijn nog steeds veel soorten die momenteel niet rechtstreeks in de UCSC Genome Browser kunnen worden opgevraagd.
De identificatie van geconserveerde domeinen of sequentiekenmerken binnen geïdentificeerde microproteïnen kan helpen het vertrouwen in hun functionele relevantie te vergroten en enig inzicht te geven in hun vermeende functie. Hier geven we aanbevelingen voor specifieke hulpmiddelen en middelen die kunnen worden gebruikt om geïdentificeerde microproteïne aminozuursequenties in meer detail te analyseren om dergelijk inzicht te krijgen. De specifieke hulpmiddelen die hieronder worden vermeld (en samengevat in de tabel met materialen) zijn vrij beschikbaar voor het publiek en we hebben ontdekt dat ze bijzonder gebruiksvriendelijk en robuust zijn in microproteïnestudies 18,38,39,40,41,47. Naast de tools die hier worden beschreven, zijn er een groot aantal extra bronnen die te vinden zijn in bioinformatica resource portals zoals Expasy (https://www.expasy.org) en EMBL-EBI (https://www.ebi.ac.uk/services/all). Het detailleren van de details voor elk van de tools binnen deze repositories valt echter buiten het bestek van dit artikel. Hier raden we de volgende bronnen aan.
Ten eerste analyseert TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) eiwitsequenties die van belang zijn voor de aanwezigheid van transmembraandomeinen. Met name een aantal microproteïnen die tot nu toe functioneel zijn gekarakteriseerd, bevatten single-pass transmembraandomeinen, wat hun lokalisatie naar membraangebieden vergemakkelijkt en hun directe regulatie van ionkanalen, wisselaars en membraangeassocieerde enzymen mogelijk maakt30. Ten tweede is het National Center for Biotechnology Information (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) een populair hulpmiddel dat wordt gebruikt om geconserveerde domeinen binnen eiwit- of coderende nucleotidesequenties te identificeren. Ten derde biedt de Protein family (Pfam)78-database (http://pfam.xfam.org) uitlijningen en classificaties van eiwitfamilies en -domeinen. Ten vierde is WoLF PSORT79 (https://wolfpsort.hgc.jp/) een hulpmiddel dat kan worden gebruikt om subcellulaire eiwitlokalisatie te voorspellen. Ten vijfde is COXPRESdB80 een gen-co-expressiedatabase (https://coxpresdb.jp) die co-gereguleerde genrelaties biedt om genfuncties te schatten. Ten slotte is SignalP 6.081 een veelgebruikt voorspellingsprogramma (https://services.healthtech.dtu.dk/service.php?SignalP) dat de aanwezigheid van een signaalpeptidesequentie herkent en de locatie van de splitsingsplaats voorspelt.
Samenvattend kunnen de hier beschreven methoden worden gebruikt om genomische gebieden van belang voor eiwitcoderingspotentieel effectief te analyseren met behulp van PhyloCSF op de UCSC Genome Browser. Deze methoden zijn zeer toegankelijk en kunnen gemakkelijk worden geleerd en efficiënt worden toegepast door personen zonder voorafgaande training of expertise in bio-informatica of vergelijkende genomica. Zoals hier in detail aangetoond, is PhyloCSF een krachtig hulpmiddel dat kan worden toegepast als een first-pass analyse om eiwitcoderende versus niet-coderende genen in gewervelde, ongewervelde en virale genomen te helpen onderscheiden, en de sterke punten van deze aanpak wegen zwaar op tegen de opgemerkte zwakke punten.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
De auteurs verklaren dat zij geen concurrerende financiële belangen hebben.
Acknowledgments
Dit werk werd ondersteund door subsidies van de National Institutes of Health (HL-141630 en HL-160569) en Cincinnati Children's Research Foundation (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
