

Summary
Burada açıklanan protokol, kullanıcı dostu UCSC Genom Tarayıcısında PhyloCSF kullanarak mikroprotein kodlama potansiyeli için ilgilenilen genomik bölgelerin nasıl analiz edileceğine dair ayrıntılı talimatlar sağlar. Ek olarak, varsayılan işlevleri hakkında fikir edinmek için tanımlanmış mikroproteinlerin dizi özelliklerini daha fazla araştırmak için çeşitli araçlar ve kaynaklar önerilmektedir.
Abstract
Yeni nesil dizileme (NGS), genomik alanını ileriye taşıdı ve çok sayıda hayvan türü ve model organizma için tüm genom dizileri üretti. Bununla birlikte, bu dizi bilgisinin zenginliğine rağmen, kapsamlı gen ek açıklama çabalarının, özellikle küçük proteinler için zorlayıcı olduğu kanıtlanmıştır. Özellikle, geleneksel protein ek açıklama yöntemleri, genom boyunca katlanarak daha fazla sayıda sahte kodlamayan sORF'yi filtrelemek için 300 nükleotitten daha kısa açık okuma çerçeveleri (sORF'ler) tarafından kodlanan varsayılan proteinleri kasıtlı olarak dışlamak için tasarlanmıştır. Sonuç olarak, mikroproteinler (<100 amino asit uzunluğunda) olarak adlandırılan yüzlerce fonksiyonel küçük protein, yanlış kodlamayan RNA'lar olarak sınıflandırılmış veya tamamen göz ardı edilmiştir.
Burada, evrimsel korumaya dayalı mikroprotein kodlama potansiyeli için genomik bölgeleri sorgulamak üzere ücretsiz, halka açık biyoinformatik araçlardan yararlanmak için ayrıntılı bir protokol sunuyoruz. Özellikle, kullanıcı dostu California Santa Cruz Üniversitesi (UCSC) Genom Tarayıcısında Filogenetik Kodon İkame Frekansları (PhyloCSF) kullanarak dizi korunumu ve kodlama potansiyelinin nasıl inceleneceğine dair adım adım talimatlar sunuyoruz. Ek olarak, amino asit dizisi korumasını görselleştirmek için tanımlanmış mikroprotein dizilerinin çoklu tür hizalamalarını verimli bir şekilde oluşturmak için adımları detaylandırıyoruz ve tahmin edilen etki alanı yapıları da dahil olmak üzere mikroprotein özelliklerini analiz etmek için kaynaklar öneriyoruz. Bu güçlü araçlar, kanonik olmayan genomik bölgelerdeki varsayılan mikroprotein kodlama dizilerini tanımlamaya yardımcı olmak veya ilgilenilen kodlamayan bir transkriptte translasyonel potansiyele sahip korunmuş bir kodlama dizisinin varlığını dışlamak için kullanılabilir.
Introduction
Genomdaki tüm kodlama elemanlarının tanımlanması, İnsan Genomu Projesi'nin başlatılmasından bu yana ana hedef olmuştur ve biyolojik sistemlerin ve genetik temelli hastalıkların etiyolojisinin anlaşılmasına yönelik merkezi bir hedef olmaya devam etmektedir 1,2,3,4. NGS tekniklerindeki ilerlemeler, omurgalılar, omurgasızlar, maya ve bitkiler de dahil olmak üzere çok sayıda organizma için tüm genom dizilerinin üretilmesine yol açmıştır5. Ek olarak, yüksek verimli transkripsiyonel dizileme yöntemleri, hücresel transkriptomun karmaşıklığını daha da ortaya çıkarmış ve hem protein kodlayan hem de kodlamayan işlevlere sahip binlerce yeni RNA molekülünü tanımlamıştır 6,7. Bu büyük miktardaki dizi bilgisinin kodunun çözülmesi devam eden bir süreçtir ve kapsamlı gen ek açıklama çabaları ile zorluklar devam etmektedir8.
Ribozom profillemesi 9,10 ve poli-ribozom dizilimi 11 de dahil olmak üzere translasyonelprofilleme yöntemlerinin son zamanlarda geliştirilmesi, yüzlerce kanonik olmayan çeviri olayının, mikroproteinler veya mikropeptitler 12,13,14,15,16 olarak adlandırılan küçük proteinler üretme potansiyeline sahip, genom boyunca şu anda açıklamasız sORF'lerle eşleştiğini gösteren kanıtlar sağlamıştır. 17. Mikroproteinler, küçük boyutları (<100 amino asit) ve klasik protein kodlayan gen özelliklerinin eksikliği nedeniyle standart gen ek açıklama yöntemleri tarafından daha önce göz ardı edilen çok yönlü proteinlerin yeni bir sınıfı olarak ortaya çıkmıştır 8,12,18,19,20. Mikroproteinler, maya21,22, sinekler 17,23,24 ve memeliler 25,26,27,28 dahil olmak üzere hemen hemen tüm organizmalarda tanımlanmıştır ve gelişim, metabolizma ve stres sinyalizasyonu dahil olmak üzere çeşitli süreçlerde kritik roller oynadığı gösterilmiştir19,20,29, 30,31,32,33,34. Bu nedenle, uzun zamandır göz ardı edilen bu fonksiyonel küçük protein sınıfının ek üyeleri için genom madenciliğine devam etmek zorunludur.
Mikroproteinlerin biyolojik öneminin yaygın olarak tanınmasına rağmen, bu gen sınıfı genom ek açıklamalarında çok az temsil edilmeye devam etmektedir ve doğru tanımlamaları, alandaki ilerlemeyi engelleyen devam eden bir zorluk olmaya devam etmektedir. Mikroprotein kodlama dizilerinin tanımlanmasıyla ilgili zorlukların üstesinden gelmek için son zamanlarda çeşitli hesaplama araçları ve deneysel yöntemler geliştirilmiştir (birkaç kapsamlı derlemede kapsamlı bir şekilde tartışılmıştır 8,35,36,37). Birçok yeni mikroprotein tanımlama çalışması 38,39,40,41,42,43,44,45,46,47, PhyloCSF 48,49 adı verilen böyle bir algoritmanın kullanımına büyük ölçüde güvenmiştir. , genomun korunmuş protein kodlayan bölgelerini kodlamayanlardan ayırt etmek için kullanılabilecek güçlü bir karşılaştırmalı genomik yaklaşım.
PhyloCSF, protein kodlayan genlerin evrimsel imzalarını tespit etmek için çok türlü nükleotid hizalamalarını ve filogenetik modelleri kullanarak kodon ikame frekanslarını (CSF) karşılaştırır. Bu ampirik model tabanlı yaklaşım, proteinlerin öncelikle nükleotid dizisinden ziyade amino asit seviyesinde korunduğu öncülüne dayanır. Bu nedenle, aynı amino asidi kodlayan eş anlamlı kodon ikameleri veya korunmuş özelliklere (yani yük, hidrofobiklik, polarite) sahip amino asitlere kodon ikameleri pozitif olarak puanlanırken, yanlış ve saçma sapan ikameler de dahil olmak üzere eş anlamlı olmayan ikameler negatif puan alır. PhyloCSF, tüm genom verileri üzerinde eğitilmiştir ve mikroproteinleri veya standart protein kodlayan genlerin bireysel ekzonlarını analiz ederken gerekli olan tam diziden izole edilmiş bir kodlama dizisinin (CDS) kısa bölümlerinin puanlanmasında etkili olduğu kanıtlanmıştır48,49.
Özellikle, California Santa Cruz Üniversitesi (UCSC) Genom Tarayıcısı 49,50,51'deki PhyloCSF izleme merkezlerinin yakın zamanda entegrasyonu, tüm geçmişlerden araştırmacıların, protein kodlama potansiyeli için ilgilenilen genomik bölgeleri sorgulamak için kullanıcı dostu bir arayüze kolayca erişmelerini sağlar. Aşağıda özetlenen protokol, PhyloCSF izleme merkezlerinin UCSC Genom Tarayıcısına nasıl yükleneceği ve daha sonra yüksek güvenilirlikli protein kodlama bölgelerini (veya bunların eksikliğini) araştırmak için ilgili genomik bölgelerin nasıl sorgulanacağı konusunda ayrıntılı talimatlar sağlar. Ek olarak, pozitif bir PhyloCSF skorunun gözlenmesi durumunda, mikroprotein kodlama potansiyelini daha fazla analiz etmek ve türler arası dizi korumasını göstermek için tanımlanmış amino asit dizilerinin çoklu tür hizalamalarını verimli bir şekilde üretmek için adımlar tanımlanmıştır. Son olarak, tartışmada, öngörülen etki alanı yapıları ve varsayılan mikroprotein fonksiyonuna ilişkin içgörü de dahil olmak üzere, tanımlanmış mikroprotein özelliklerini araştırmak için halka açık birkaç ek kaynak ve araç tanıtılmıştır.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Aşağıda özetlenen protokol, UCSC Genom Tarayıcısına (Mudge ve ark.49 tarafından oluşturulan) PhyloCSF tarayıcı izlerini yükleme ve gezinme adımlarını ayrıntılarıyla açıklamaktadır. UCSC Genom Tarayıcısı ile ilgili genel sorular için, kapsamlı bir Genom Tarayıcısı Kullanım Kılavuzu burada bulunabilir: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. PhyloCSF İzleme Hub'ını UCSC Genom Tarayıcısına Yükleme
- Bir internet tarayıcısı penceresi açın ve UCSC Genom Tarayıcısı'na (https://genome.ucsc.edu/) gidin.
- Araçlarımız başlığı altında, Hub'ları İzle seçeneğini belirleyin.
NOT: Hub'ları İzle seçeneği, Verilerim sekmesi altında da bulunabilir. - Genel Hub'lar sekmesinde, Arama terimleri kutusuna PhyloCSF yazın. Genel Hub'larda Ara düğmesine tıklayın.
- Hub Adı PhyloCSF (Açıklama: PhyloCSF tarafından ölçülen evrimsel protein kodlama potansiyeli) için Bağlan düğmesine tıklayarak PhyloCSF'ye bağlanın.
NOT: Bu İzleme Hub'ı, insan (hg19 ve hg38) ve fare (mm10 ve mm39) dahil olmak üzere çok sayıda montaja yüklenir. - Bağlan'a tıkladıktan sonra, UCSC Genome Tarayıcı Ağ Geçidi sayfasına (https://genome.ucsc.edu/cgi-bin/hgGateway) yönlendirilmeyi bekleyin.
2. Gen Tanımlayıcıları kullanarak ilgilenilen genlere gitme
- Sorgulanacak türü ve genom derlemesini seçin. Farklı bir türü (ör. fare) sorgulamak için, uygun simgeye tıklayarak Gözat/Tür Seç başlığı altında ilgilendiğiniz türü seçin veya Tür, ortak ad veya derleme kimliği girin yazan metin kutusuna türü yazın.
NOT: Derleme doğrudan Konum Bul başlığı altında listelenir. Tipik olarak, varsayılan İnsan Meclisi'dir (örneğin, Aralık 2009 [GRCh37/hg19]). - Açılır menüyü kullanarak Konum Bul başlığı altında aranacak montajı seçin.
- Konum/Arama Terimi kutusuna konumu, gen sembolünü veya arama terimlerini girin ve Genom Tarayıcısında ilgilenilen bir gene gitmek için Git'e tıklayın.
- Arama birden fazla eşleşmeyle sonuçlandıysa, ilgilenilen bir konumun seçilmesini gerektiren bir sayfaya yönlendirilmeyi bekleyin. İlgilendiğiniz uygun gene tıklayın.
3. Dizi bilgilerini kullanarak ilgilenilen genomik bölgelere gitme
- UCSC Genom Tarayıcısına (https://genome.ucsc.edu/) gidin ve belirli bir DNA veya protein dizisini sorgulamak için Araçlarımız başlığı altındaki BLAST Benzeri Hizalama Aracı'nı (BLAT) seçin. Alternatif olarak, imleci Araçlar sekmesinin üzerine getirin ve Blat seçeneğini belirleyin veya şu bağlantıyı izleyin: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Açılır menüleri kullanarak ilgilenilen türü (Genom) ve Derlemeyi seçin.
- Açılır menüyü kullanarak Sorgu türünü tanımlayın.
- İlgilendiğiniz sırayı BLAT Arama Genomu metin kutusuna yapıştırın ve Gönder'i tıklayın.
- İlgilenilen genomik bölgeye gitmek için EYLEMLER başlığı altındaki tarayıcı bağlantısına tıklayın.
4. PhyloCSF İzleme Verileri kullanarak korunmuş sORF'lerin tanımlanması
- PhyloCSF bölgelerinin pozitif skorlanması için genomik ilgi alanını görsel olarak tarayın (Şekil 1).
NOT: UCSC Genom Tarayıcısında PhyloCSF skorlarının görsel olarak nasıl yorumlanacağına ilişkin ayrıntılı bir açıklama için, aşağıdaki temsili sonuçlar bölümüne bakın. - Dizi özelliklerini incelemek ve başlatma/durdurma kodonlarını aramak üzere ilgilenilen bölgeleri büyütmek için yakınlaştırma özelliğini kullanın. Manuel olarak yakınlaştırmak için, shift tuşunu basılı tutun ve ilgilenilen bölge boyunca sürüklerken fare düğmesini tıklayıp basılı tutun. Alternatif olarak, gezinmek için sayfanın üst kısmındaki yakınlaştırma ve uzaklaştırma düğmelerini kullanın (1,5x, 3x, 10x veya temel yakınlaştırma seçenekleri kullanılabilir).
NOT: Yakınlaştırma/uzaklaştırma düğmelerini kullanmadan önce, geni ilgi alanı ekranın ortasında olacak şekilde yeniden konumlandırmak gerekir. Bu eylemi gerçekleştirmek için, resme tıklayın ve genomik bölgeyi istediğiniz gibi yatay olarak taşımak için sola veya sağa sürükleyin veya sayfanın üst kısmındaki hareket oklarını kullanın. - Nükleotid (baz) dizisi görünene kadar yakınlaştırın.
NOT: Nükleotid dizisi doğrudan +1 Düzleştirilmiş FiloCSF skorunun üzerinde görünecektir. - Varsayılan başlangıç (ATG) ve durdurma (TGA / TAA / TAG) kodonlarını tanımlamak için pozitif skorlama yapan PhyloCSF bölgelerinin başlangıcına ve sonuna yakın nükleotid dizisini görsel olarak tarayın.
NOT: Eğer ilgilenilen gen DNA'nın eksi ipliği üzerindeyse, başlangıç ve bitiş kodonları ters tamamlayıcı olacaktır (yani, başlangıç kodonu için CAT ve durdurma kodonu için TCA / TTA / CTA).
5. Diğer genomlardaki homolog bölgelerin görüntülenmesi
- Fareyi sayfanın üst kısmındaki Görünüm başlığının üzerine getirin ve Diğer Genomlarda (Dönüştür) seçeneğine tıklayın.
- Yeni Genom başlığının altındaki açılır menüyü kullanarak ilgilendiğiniz genomu tanımlayın.
- Yeni Montaj başlığının altındaki açılır menüyü kullanarak ilgilendiğiniz genomik derlemeyi seçin ve ardından Gönder düğmesini tıklayın.
- Tarayıcı, yeni derlemedeki benzer bölgelerin bir listesini döndürdüğünde, ilgilenilen homolog bölgeye gitmek için kromozom konumu bağlantısına tıklayın.
NOT: Toplam bazların (nükleotitler) yüzdesi ve bölgenin kapsadığı açıklık, listelenen her bölge için tanımlanacaktır. Eşleşen bazların yüzdesi ne kadar yüksek olursa, koruma ilgilenilen bölge için o kadar yüksek olur. - Diziyi analiz etmek için Bölüm 4'te ayrıntılı olarak açıklanan aynı navigasyon stratejilerini izleyin.
6. İlgilenilen mikroproteinler için çok türlü dizi hizalamalarının oluşturulması
- Gen açıklama sayfasına gitmek için UCSC Genom Tarayıcısındaki GENCODE parçasında (Şekil 1A'da mavi kutuyla gösterilmiştir) ilgilenilen gene tıklayın.
- Sıralı ve Araçlara ve Veritabanlarına Bağlantılar başlığı altında, tablodaki Diğer Türler FASTA yazan bağlantıya tıklayın.
- Seçmek için ilgilenilen türlerle ilişkili kutulara tıklayın. Gönder'e tıklayın. Sayfanın altında görünen dizileri kopyalayıp FASTA biçiminde bir sözcük işlem belgesine yapıştırın.
- İkinci bir tarayıcı penceresi açın ve Avrupa Biyoinformatik Enstitüsü (EMBL-EBI) web sitesi 53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/'deki Clustal Omega Çoklu Dizi Hizalama aracı 52'ye gidin.
- Panoda hala bulunan dizi dosyalarını, dizileri desteklenen herhangi bir biçimde okuyan ADIM 1'deki kutuya yapıştırın. Sayfanın en altına gidin ve Gönder'i tıklayın. Her amino asidin korunum derecesini gösteren semboller için hizalanmış sonuçların (siyah yazı tipiyle) altına bakın (semboller Tablo 1'de tanımlanmıştır).
NOT: Hizalamanın oluşturulması birkaç dakika sürebilir. - Amino asit özelliklerini renkli olarak görüntülemek için, amino asitleri özelliklerine göre renklendirmek için dizilerin hemen üzerindeki Renkleri Göster bağlantısına tıklayın ( Tablo 2'de tanımlanmıştır).
- Bir şekil veya illüstrasyon dosyası oluşturmak için dizi hizalamasını kopyalayıp bir sözcük işlem veya slayt gösterisi programına yapıştırın (örneğin, Şekil 2).
NOT: Hizalama için Courier gibi tek aralıklı bir yazı tipi kullanın. - Clustal Omega sonuç sayfasındaki diğer çıktıları görüntülemek için uygun sekmelere tıklayın (örneğin, Kılavuz Ağacı veya Filogenetik Ağaç).
- Birden fazla dizi hizalaması düzenleme, görselleştirme ve analiz55 konusunda uzmanlaşmış ücretsiz bir program olan Jalview'i kullanarak dizi bilgilerini görüntüleme veya MView ve Simple Phylogeny56'ya doğrudan bağlantılara erişme seçenekleri için Sonuç Görüntüleyiciler sekmesine tıklayın.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Burada, korunmuş bir sORF'nin UCSC Genom Tarayıcısında kolayca görselleştirilebilen ve analiz edilebilen pozitif bir PhyloCSF skorunu nasıl üreteceğini göstermek için doğrulanmış mikroprotein mitoregulini (Mtln) örnek olarak kullanacağız. Mitoregulin daha önce kodlamayan bir RNA (eski adıyla insan gen kimliği LINC00116 ve fare gen kimliği 1500011K16Rik) olarak açıklanmıştı. Karşılaştırmalı genomik ve dizi koruma analizi yöntemleri, ilk keşfinde kritik bir rol oynamıştır 40,57,58,59,60,61 ve bu yöntemlerin gücünü vurgulamaktadır. Bu örnekte, fare GRCm38/mm10 (Aralık 2011) montajı kullanılacaktır. Arama, protokol bölüm 2'de açıklandığı gibi gen tanımlayıcıları (mitoregulin, Mtln) veya gen pozisyonu (chr2: 127.791.364-127.792.496) kullanılarak gerçekleştirilebilir. Alternatif olarak, mitoregulin için amino asit dizisi (Şekil 2'de gösterilmiştir) BLAT aracı kullanılarak aranabilir (protokol bölüm 3'te açıklanmıştır).
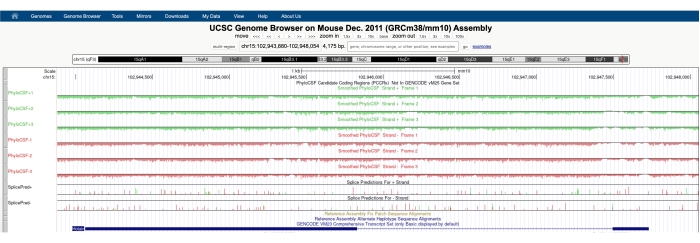
Şekil 1A'da gösterilene benzer bir ekran, ekranın üst kısmında görünen PhyloCSF Track Hub ile birlikte görünecektir. Düzleştirilmiş FiloCSF izleri (her bir kodonun kodlama olasılığını tanımlayan gizli bir Markov modeli ile düzeltilmiş), DNA'nın artı ipliğine karşılık gelen üç parça (yeşil renkte PhyloCSF +1, +2 ve +3 olarak tasvir edilmiştir) ve DNA'nın eksi ipliğine karşılık gelen üç iz (kırmızı renkte PhyloCSF -1 olarak tasvir edilmiştir) ile altı toplam iz olarak tasvir edilmiştir. -2 ve -3). Bu izler, her yöne ilgi duyan gen için üç potansiyel okuma çerçevesini temsil eder. Tarayıcı penceresinde, ekzonlar, intronları temsil eden ince mavi yatay çizgilerle bağlanmış mavi dikdörtgenler olarak tasvir edilir. İntronik bölgelerdeki ok uçları, genin hangi yönde yazıldığını (ve dolayısıyla PhyloCSF skoru için hangi ipliğe odaklanılacağını) gösterir. Şekil 1'deki Mtln örneğinde, intronic ok uçları sola işaret ediyor. Bu nedenle, Mtln geni DNA'nın eksi ipliğinden kopyalanır ve ilgili PhyloCSF skoru -1, -2 ve -3 izlerinde (kırmızı) tasvir edilir.
Her PhyloCSF izi, çizginin altında açık yeşil / kırmızı renkte gösterilen negatif puanlama bölgeleri ve çizginin üstünde koyu yeşil / kırmızı ile gösterilen pozitif puanlama bölgeleri ile ince siyah bir çizgi olarak tasvir edilmiştir. Girişte açıklandığı gibi, pozitif bir PhyloCSF skoru, muhtemelen kodlama olan korunmuş bir bölgeyi gösterir. Özellikle yüksek sekans korunumuna sahip protein kodlayan bölgeler için, genellikle antisens ipliği üzerinde de pozitif puan aldıklarını unutmayın; Bununla birlikte, PhyloCSF skoru genellikle doğru iplikçikte daha yüksektir. Örneğin, bu, doğru kodlama dizisinin PhyloCSF -1 pistinde çok yüksek puan aldığı Mtln için Şekil 1'de görülebilir ve antisens ipliği (PhyloCSF +2 izi) de pozitif bir skor üretir. Şekil 1A'da görüldüğü gibi (kara kutu ile gösterilmiştir), Mtln'nin ilk ekzonunda PhyloCSF -1 yolunda çok yüksek puan alan bir bölge vardır, bu da bunun bir kodlama bölgesine karşılık gelebileceğini düşündürmektedir. Bu bölgeyi daha ayrıntılı incelemek için, bölgeyi yakınlaştırmak ve büyütmek yararlı olacaktır (Şekil 1B). Şekil 1C, D'de gösterildiği gibi, Mtln'nin ilk ekzonundaki pozitif puanlama bölgesi doğrudan bir başlangıç kodonu (Şekil 1C) üzerinden başlar ve bu ORF'nin yüksek oranda korunduğunu gösteren ve güçlü bir şekilde kodlayıcı bir ORF olduğunu gösteren bir durdurma kodonunda (Şekil 1D) sona erer. Mtln, DNA'nın eksi ipliğinde olduğu için, başlangıç ve bitiş kodonları kodonun ters tamamlayıcısı olarak gösterilir (yani, ATG başlangıç kodonu CAT [Şekil 1C] ve TGA durdurma kodonu TCA [Şekil 1D] olarak gösterilir).
Mikroprotein kodlama potansiyeline sahip korunmuş bölgeleri aramak için PhyloCSF'yi kullanmanın yanı sıra, bu teknik, korunmuş bir ORF'nin varlığını dışlamak için varsayılan kodlamayan RNA'ların ilk geçiş analizi olarak da uygulanabilir, böylece kodlamayan bir ek açıklama için destek sağlar. Örneğin, iyi karakterize edilmiş lncRNA HOTAIR62,63'ün PhyloCSF kullanılarak analizi, altı parçanın tamamında tüm gen boyunca negatif bir skor göstermektedir (Şekil 3), sekans korunumu eksikliğini güçlü bir şekilde göstermektedir ve HOTAIR'in kodlamayan bir RNA olarak doğru bir şekilde açıklandığına dair destek sağlamaktadır.
Şekil 1'de açıkça görüldüğü gibi, mitoregulin için tüm kodlama ORF'si tek bir ekzon içinde bulunur, böylece PhyloCSF tarafından tek, kesintisiz, pozitif puanlama bölgesi ile basit ve anlaşılır bir okuma üretilir. Bununla birlikte, PhyloCSF izleme göbeği verileri her zaman net ve yorumlanması kolay değildir. Örneğin, fare tarafından kodlanan mitolamban/Stmp1/Mm47 mikroproteini 1810058I24Rik geni 47,64,65, üç ekzonu kapsayan korunmuş bir ORF'yi tasvir eder (Şekil 4A) ve pozitif PhyloCSF skoru ekzon 1'deki +2 izinden (Şekil 4B) ekzon 2'deki +3 izine (Şekil 4C) ve ardından ekzon 3'teki +2 izine geri döner (Şekil 4D) ). İlk bakışta bu kafa karıştırıcı görünse de, açıklama oldukça basittir. PhyloCSF, genomik bölgelerin altı potansiyel okuma çerçevesini (üçü DNA'nın artı ipliğinde ve üçü eksi iplikçikte) her gen için spesifik ekzon / intron mimarisini dikkate almadan puanlar. Bu nedenle, okuma çerçevelerinin 3-nükleotid periyodikliğindeki intronik dizi bilgisini korur. Bu nedenle, bir intron üçe bölünemeyen bir dizi nükleotid içeriyorsa (yani, üç nükleotid / kodon), PhyloCSF okuma çerçevesi bir parçadan diğerine atlayacaktır.
Son olarak, PhyloCSF, tek bir RNA molekülü içindeki birden fazla farklı kodlayan ORF'yi tanımlamak için de etkili bir şekilde kullanılabilir. Örneğin, MIEF1 mikroproteini (MIEF1-MP), mitokondriyal uzama faktörü 1'in (MIEF1)66'sının 5' UTR'si içinde kodlanmıştır (Şekil 5). MIEF1 genomik bölgesi PhyloCSF tarafından analiz edildiğinde, MIEF1-MP'ye karşılık gelen ayrık bir pozitif PhyloCSF skoru (Şekil 5C), MIEF1 için ana CDS'nin yukarı akışında kolayca gözlemlenebilir (Şekil 5B). MIEF1 ve ilişkili mikroprotein (MIEF1-MP) hakkında daha fazla tartışma, bu makalede özetlenen yöntem ve protokollerin güçlü ve zayıf yönlerinin bir özeti ile birlikte aşağıda verilmiştir.

Şekil 1: Mitoregulin (Mtln) geninin filoCSF analizi, doğrulanmış bir mikroproteine karşılık gelen yüksek sekanslı bir korunum bölgesini gösterir. (A) UCSC Genom Tarayıcısı ve PhyloCSF İzlerinin ekran görüntüleri, Mtln'nin iki ekzon ve tek bir intron içerdiğini göstermektedir. İntrondaki ok uçları sola işaret eder, Mtln geninin DNA'nın eksi ipliğinden kopyalandığını gösterir ve bu nedenle ilgili PhyloCSF skorları -1, -2 ve -3 izlerinde (kırmızı) gösterilir. Tam mitoregulin kodlama dizisi Exon 1'de bulunur ve PhyloCSF -1 pistinde (B) yüksek puan alır. Yeşil bir kutu (CAT, ters kompleman ATG) ile vurgulanan PhyloCSF -1 pistindeki (C) pozitif puanlama bölgesinin başlangıcında korunmuş bir başlangıç kodonu açıkça gözlemlenebilir. Ek olarak, korunmuş bir durdurma kodonu (TCA, ters kompleman TGA), pozitif skorlama yapan PhyloCSF bölgesinin ucuyla aynı hizada olan panelde (D) kırmızı bir kutu ile gösterilir. Mtln geni hakkında ayrıntılı bilgi, mavi kutunun içindeki Mtln gen tanımlayıcısına tıklayarak bulunabilir (panel A'da gösterilmiştir). Not olarak, yüksek oranda korunmuş protein kodlayan bölgeler genellikle antisens ipliğinde de pozitif puan alır (burada Mtln için PhyloCSF +2 pistinde görülür). Bununla birlikte, PhyloCSF skoru tipik olarak doğru iplikçikte daha yüksektir (bu örnekte PhyloCSF -1 izi). Bu şeklin daha büyük bir versiyonunu görüntülemek için lütfen buraya tıklayın.

Şekil 2: Clustal Omega programı kullanılarak üretilen mikroprotein mitoregulinin çoklu tür dizisi hizalaması. Belirtilen sekiz tür için mitoregulin amino asit dizileri, protokol bölüm 6'da ayrıntılı olarak açıklandığı gibi ekstrakte edildi ve Clustal Omega çoklu dizi hizalama aracı ile hizalandı. Amino asitlerin özellikleri renkle gösterilir (kırmızı, küçük / hidrofobik; mavi, asidik; macenta, bazik; yeşil, hidroxl / sülfhidril / amin) ( Tablo 2'de daha ayrıntılı olarak tanımlanmıştır). Amino asitlerin altındaki semboller korunumun derecesini gösterir (yıldızlar, tam olarak korunmuş kalıntılar; kolonlar, güçlü bir şekilde benzer özelliklere sahip amino asitler; dönemler, zayıf benzer özelliklere sahip gruplar arasındaki korunum) ( Tablo 1'de kapsamlı bir şekilde detaylandırılmıştır). Bu şeklin daha büyük bir versiyonunu görüntülemek için lütfen buraya tıklayın.
Şekil 3: Doğrulanmış uzun kodlamayan RNA Hotair için PhyloCSF izlerinin ekran görüntüsü, genomik lokusu boyunca dizi korunumu eksikliğini göstermektedir. Hotair'in intronik bölgesindeki ok uçları sola işaret ediyor, bu da lncRNA'nın DNA'nın negatif ipliğinden transkribe edildiğini ve bu nedenle PhyloCSF -1, -2 ve -3 izlerinin analizin odağı olması gerektiğini gösteriyor. PhyloCSF skorunun tüm gen boyunca negatif olduğunu (altı parçanın tümü için), kodlamayan bir RNA olarak uygun ek açıklamasını destekleyen dizi korunumunun eksikliğini gösterdiğini unutmayın. Bu şeklin daha büyük bir versiyonunu görüntülemek için lütfen buraya tıklayın.

Şekil 4: Mikroprotein mitolamban/Stmp1/Mm47'yi kodlayan fare 1810058I24Rik geninin filoCSF analizi. (A) Fare 1810058I24Rik geni üç ekzondan oluşur ve intronik bölgelerdeki ok uçları sağa işaret eder, bu da DNA'nın artı ipliğine yazıldığını ve bu nedenle PhyloCSF +1, +2 ve +3 izlerinin analiz edilmesi gerektiğini gösterir. Korunmuş mikroprotein kodlama dizisi, ekzon 1 (B)'den başlayarak, ekzon 2'den (C) geçerek ve ekzon 3'te (D) biten üç ekzonu da kapsar. Pozitif PhyloCSF puanının ekzon 1'deki +2 pistinde, ekzon 2'deki +3 pistinde ve ekzon 1'deki +2 pistinde bulunduğunu unutmayın. Pozitif skorun bir parçadan diğerine hareketinin nedeni, PhyloCSF'nin genin ekzon / intron yapısından bağımsız olarak DNA dizisinin altı potansiyel okuma çerçevesini analiz etmesidir. Bu nedenle, üç (üç nükleotid / kodon) bölünemeyen bir dizi nükleotid içeren bir intron, okuma çerçevesinde farklı bir parçaya kaymaya neden olacaktır. Bu şeklin daha büyük bir versiyonunu görüntülemek için lütfen buraya tıklayın.

Şekil 5: Mief1 genomik lokusunun PhyloCSF ile analizi, paylaşılan RNA üzerindeki ana Mief1 CDS'sinden bağımsız olan 5' UTR'de protein kodlama potansiyeline sahip bir bölgeyi tanımlar. Bu korunmuş yukarı akış ORF'sinin (uORF) Mief1-MP adlı bir mikroproteini kodladığı gösterilmiştir. (A) Mief1 genomik lokusuna genel bakış. İntronlardaki ok uçları sağa işaret eder ve Mief1'in DNA'nın artı ipliğinden kopyalandığını gösterir (kodlama potansiyelini belirlemek için PhyloCSF +1, +2 ve +3 izlerine odaklanın). Ana Mief1 CDS, 463 amino asit proteinini kodlar ve panelde (B) gösterilir. Bununla birlikte, Mief1'in 5' UTR'sinde, Mief1-MP (C) adı verilen benzersiz bir 70 amino asit mikroproteinini kodlayan ayrı bir korunmuş yukarı akış ORF da vardır. Panel C'de görüldüğü gibi, Mief1-MP, Mief1 5' UTR içinde kendi korunmuş başlangıç ve bitiş kodonuna sahiptir ve ORF, PhyloCSF +1 pistinde çok yüksek puan alır ve fonksiyonel bir mikroproteini kodladığına dair güçlü kanıtlar sağlar. Kısaltmalar: ORF = açık okuma çerçevesi; uORF = yukarı akış ORF; UTR = çevrilmemiş bölge; CDS = kodlama sırası. Bu şeklin daha büyük bir versiyonunu görüntülemek için lütfen buraya tıklayın.
| Sembol | Amino Asit Koruma Seviyesi | Gruplandırılmış Amino Asitler |
| Yıldız işareti (*) | Tamamen korunmuş kalıntı | Uygulanamaz (tek, tamamen korunmuş kalıntı) |
| Kolon (:) | Çok benzer özelliklere sahip gruplar | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; cesaret |
| Dönem (.) | Zayıf benzer özelliklere sahip gruplar | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; cesaret |
| Boşluk (sembol yok) | Benzerlik yok | Uygulanamaz (benzerlik yok) |
Tablo 1: Clustal Omega tarafından oluşturulan Çoklu Dizi Hizalamaları için konsensüs sembollerinin tanımları. Şekil 2'de gösterilen çoklu tür dizisi hizalaması, Clustal Omega52 kullanılarak oluşturulmuştur. Kısaltmalar: serin (S), treonin (T), alanin (A), asparajin (N), glutamik asit (E), glutamin (Q), lizin (K), aspartik asit (D), arginin (R), metiyonin (M), izolösin (I), lösin (L), fenilalanin (F), histidin (H), tirozin (Y), triptofan (W), sistein (C), valin (V), glisin (G), prolin (P).
| Font Rengi | Mülk | Amino Asit Kalıntısı [Kısaltma] |
| Kırmızı | Küçük, hidrofobik | alanin [A], valin [V], fenilalanin [F], prolin [P], metiyonin [M], izolösin [I], lösin [L], triptofan [W] |
| Mavi | Asidik | aspartik asit [D], glutamik asit [E] |
| Galibarda | Temel | arginin [ R ], lizin [ K ] |
| Yeşil | Hidroxl, sülfhidril, amin, +G | serin [S], treonin [T], tirozin [Y], histidin [H], sistein [C], asparajin [N], glisin [G], glutamin [Q] |
Tablo 2: Şekil 2'de gösterilen amino asitlerin özellikleri. Clustal Omega52 , Şekil 2'de gösterilen çoklu dizi hizalamasını oluşturmak için kullanıldı.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Burada sunulan protokol, kullanıcı dostu UCSC Genom Tarayıcısı48,49,50,51'de PhyloCSF kullanarak mikroprotein kodlama potansiyeli için ilgilenilen genomik bölgelerin nasıl sorgulanacağına dair ayrıntılı talimatlar sunmaktadır. Yukarıda ayrıntılı olarak açıklandığı gibi, PhyloCSF, protein kodlayan genlerin tipik48,49 evrimsel imzalarını tanımlamak için filogenetik modelleri ve kodon ikame frekanslarını birleştiren güçlü bir karşılaştırmalı genomik algoritmadır. PhyloCSF, daha önce kodlamayan 38,39,40,41,42,43,44,45,46,47 olarak belirtilen genomik bölgelerdeki fonksiyonel mikroproteinleri tanımlamak için yaygın olarak kullanılmaktadır. ve bu yaklaşımın, 13 amino asit kadar küçük mikroproteinler ve kanonik proteinlerin küçük ekzonları 35,48,49 gibi kısa diziler için diğer karşılaştırmalı genomik yöntemlerden daha iyi performans gösterdiği gösterilmiştir. Özellikle, PhyloCSF'nin evrimsel koruma yoluyla fonksiyonel protein kodlama dizilerini tanımlamak için sağlam bir yöntem olarak faydası, omurgalı ve omurgasız türlerin ötesine uzanır ve hatta SARS-CoV-2 genomunun protein kodlama kapasitesini başarılı bir şekilde sorgulamak için viral genomlara uygulanmıştır67.
Açıklamalı kodlamayan RNA'lar içindeki varsayılan kodlama dizilerini tanımlamanın yanı sıra, PhyloCSF'nin bir avantajı, hem 5' yukarı akış hem de 3' aşağı akış ORF'leri (sırasıyla uORF'ler ve dORF'ler) dahil olmak üzere, kanonik protein kodlama genlerinin açıklamalı çevrilmemiş bölgelerinde (UTR'ler) ORF'ler tarafından kodlanan korunmuş mikroproteinleri güvenilir bir şekilde tespit edebilmesidir 8,19,66,68 . Örneğin, MIEF1 mikroproteini (MIEF1-MP), mitokondriyal uzama faktörü 1'in (MIEF1) 66'sının 5' UTR'sinde kodlanır. MIEF1-MP durumunda, MIEF1-MP'ye karşılık gelen ayrık pozitif bir FiloCSF skoru, MIEF1'i kodlayan ORF'nin yukarı akışı gözlenir (Şekil 5). Bazı uORF kodlu mikroproteinler, paylaşılan mRNA'larındaki aşağı akış kanonik proteinleriyle doğrudan etkileşime girerken (örneğin, MIEF1-MP ve MIEF1), diğerleri ana CDS66,68 tarafından kodlanan proteinden bağımsız olarak işlev görür. Bu nedenle, uORF kodlu mikroproteinleri karakterize ederken, aşağı akış protein ürünlerinin doğrudan düzenlenmesi yoluyla işlev gördükleri varsayılmamalıdır.
PhyloCSF, korunmuş mikroprotein kodlama dizilerinin tanımlanması için bir araç olarak birçok açık güce sahip olsa da, bu yöntemin çeşitli sınırlamalarını tanımak önemlidir. İlk olarak, dizi korunumu, bir genomik bölgenin fonksiyonel seçilimden geçtiğini ve dolayısıyla kodlama yaptığını güçlü bir şekilde öne sürerken, sağlam bir korunumun eksikliği ve bunun sonucunda ortaya çıkan negatif bir PhyloCSF skoru, belirli bir dizi için kodlama potansiyelini kesin olarak dışlamaz. Başka bir deyişle, yalnızca PhyloCSF'ye güvenmek, güçlü bir şekilde korunmayan ancak yine de fonksiyonel mikroproteinler üreten çevrilmiş ORF'lerin gözetimine neden olabilir. Özellikle, düşük korunum veya negatif koruma puanlarına sahip genomik bölgeler, türe özgü kodlama bölgelerine veya sekans ayrışması veya de novo gen doğumu46,69,70,71,72,73,74 yoluyla evrimsel "genç" genlerin bölgelerine karşılık gelebilir. Örneğin, daha önce insan kodlamayan RNA LINC00467 olduğu düşünülen şey tarafından kodlanan mikroprotein ASAP, PhyloCSF tarafından pozitif olarak puanlanmamıştır, çünkü amino asit dizisi sadece daha yüksek memelilerdekorunmaktadır 75. Ek olarak, son çalışmalar, intergenik lncRNA RP3-527G5.1 tarafından kodlanmış biri de dahil olmak üzere, pozitif bir PhyloCSF skoru68,72 üretmeyen birkaç insana özgü mikroprotein tanımlamıştır. Bu bağlamda, pozitif bir PhyloCSF skorunun olmaması, kodlamayan bir bölgenin kanıtı olarak yorumlanamaz ve dikkatle yorumlanmalıdır.
PhyloCSF kullanırken akılda tutulması gereken ikinci bir husus, pozitif bir skorun fonksiyonel seleksiyon ve protein kodlama kapasitesini oldukça düşündürmesine rağmen, bu kanıt çizgisinin tek başına duramayacağı ve deneysel olarak doğrulanması gerektiğidir. Kararlı mikroprotein ekspresyonu için destekleyici kanıtlar üretmek için kullanılabilecek yöntemlere örnek olarak, varsayılan proteinin, ilgili mikroprotein dizisine karşı yükseltilmiş bir antikor kullanılarak kütle spektrometrisi veya batı lekelenmesi ile tespiti sayılabilir. Alternatif olarak, optimal antijenite için sekans seçeneklerinin bulunmaması nedeniyle mikroproteinler için güvenilir antikorlar üretmek zor olabileceğinden, CRISPR / Cas9 ve homolojiye yönelik onarım (HDR) yolunu, varsayılan mikroprotein dizisi ile çerçevedeki endojen lokusa bir epitop etiketi sokmak için kullanmak da mümkündür, böylece yüksek afiniteli bir antikor kullanarak ilgili proteinin tespitini kolaylaştırır (örneğin, BAYRAK, HA, V5, Myc)18. PhyloCSF'nin kabul edilmesi gereken son bir sınırlaması, şu anda Homo sapiens (insan hg19, hg38), Mus musculus (fare mm10, mm39), Gallus gallus (tavuk, galGal4, galGal6), Drosophila melanogaster (meyve sineği, dm6), Caenorhabditis elegans dahil olmak üzere yaygın olarak kullanılan genomik derlemelerin çoğuna entegre edilmiş olmasına rağmen, (nematodlar, ce11) ve SARS-CoV-2 (wuhCor1), şu anda doğrudan UCSC Genom Tarayıcısında sorgulanamayan birçok tür vardır.
Tanımlanmış mikroproteinler içindeki korunmuş alanların veya dizi özelliklerinin tanımlanması, fonksiyonel alaka düzeylerine olan güveni artırmaya yardımcı olabilir ve varsayılan işlevleri hakkında bazı bilgiler sağlayabilir. Burada, bu tür bir içgörü elde etmek için tanımlanmış mikroprotein amino asit dizilerini daha ayrıntılı olarak analiz etmek için kullanılabilecek belirli araçlar ve kaynaklar için önerilerde bulunuyoruz. Aşağıda listelenen (ve Malzeme Tablosunda özetlenen) spesifik araçlar halka ücretsiz olarak sunulmaktadır ve bunları mikroprotein çalışmalarında özellikle kullanıcı dostu ve sağlam buldukları 18,38,39,40,41,47. Burada açıklanan araçların ötesinde, Expasy (https://www.expasy.org) ve EMBL-EBI (https://www.ebi.ac.uk/services/all) gibi biyoinformatik kaynak portallarında bulunabilecek çok sayıda ek kaynak vardır. Ancak, bu depolardaki araçların her birinin ayrıntılarını ayrıntılandırmak bu makalenin kapsamı dışındadır. Burada aşağıdaki kaynakları öneririz.
İlk olarak, TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0), transmembran alanlarının varlığı için ilgilenilen protein dizilerini analiz eder. Özellikle, şimdiye kadar işlevsel olarak karakterize edilen bir dizi mikroprotein, membran bölgelerine lokalizasyonlarını kolaylaştıran ve iyon kanallarının, eşanjörlerin ve membranla ilişkili enzimlerin doğrudan düzenlenmesini sağlayan tek geçişli transmembran alanları içerir30. İkincisi, Ulusal Biyoteknoloji Bilgi Merkezi (NCBI) Korunmuş Etki Alanı Arama77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), protein içindeki korunmuş alanları tanımlamak veya nükleotid dizilerini kodlamak için kullanılan popüler bir araçtır. Üçüncüsü, Protein ailesi (Pfam)78 veritabanı (http://pfam.xfam.org), protein ailelerinin ve alanlarının hizalamalarını ve sınıflandırmalarını sağlar. Dördüncüsü, WoLF PSORT79 (https://wolfpsort.hgc.jp/), hücre altı protein lokalizasyonunu tahmin etmek için kullanılabilecek bir araçtır. Beşincisi, COXPRESdB80 , gen fonksiyonlarını tahmin etmek için birlikte düzenlenmiş gen ilişkileri sağlayan bir gen ko-ekspresyon veritabanıdır (https://coxpresdb.jp). Son olarak, SignalP 6.081 , bir sinyal peptid dizisinin varlığını tanıyan ve bölünme bölgesinin yerini tahmin eden yaygın olarak kullanılan bir tahmin programıdır (https://services.healthtech.dtu.dk/service.php?SignalP).
Özetle, burada açıklanan yöntemler, UCSC Genom Tarayıcısında PhyloCSF kullanarak protein kodlama potansiyeli için ilgi çekici genomik bölgeleri etkili bir şekilde analiz etmek için kullanılabilir. Bu yöntemler oldukça erişilebilirdir ve biyoinformatik veya karşılaştırmalı genomik konusunda önceden eğitim veya uzmanlık sahibi olmayan bireyler tarafından kolayca öğrenilebilir ve verimli bir şekilde uygulanabilir. Burada ayrıntılı olarak gösterildiği gibi, PhyloCSF, omurgalı, omurgasız ve viral genomlarda protein kodlayan ve kodlamayan genleri ayırt etmeye yardımcı olmak için ilk geçiş analizi olarak uygulanabilen güçlü bir araçtır ve bu yaklaşımın güçlü yönleri, belirtilen zayıflıklardan daha ağır basmaktadır.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Yazarlar, rekabet eden finansal çıkarları olmadığını beyan ederler.
Acknowledgments
Bu çalışma, Ulusal Sağlık Enstitüleri (HL-141630 ve HL-160569) ve Cincinnati Çocuk Araştırma Vakfı (Mütevelli Ödülü) tarafından desteklenmiştir.
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
