



Overview
ソース: リアノン M. ルベケ1, ナタリア マーティン1, アンドリュー J. ヴァン アルスト1, ビクター J. ディリタ1
1ミシガン州立大学微生物・分子遺伝学科、イーストランシング、ミシガン州、アメリカ合衆国
細菌は地球上のほぼどこにでも見られる多様な微生物です。多くの特性は、グラム染色の種類、形状と配置、カプセルの生産、胞子の形成を含むが、これらに限定されない、互いにそれらを区別するのに役立ちます。これらの特性を観察するために、1つは光顕微鏡を使用することができます。しかし、いくつかの細菌特性(例えば、サイズ、着色の欠如、屈折特性)は、光顕微鏡(1、2)だけで細菌を区別することが困難になります。細菌の種類を軽い顕微鏡で区別する場合は、染色細菌が必要です。2つの主要なタイプの軽微小顕微鏡は、シンプルで化合物です。それらの主な違いは、オブジェクトを拡大するために使用されるレンズの数です。単純な顕微鏡(例えば虫眼鏡)は、物体を拡大するためのレンズが1つしかありませんが、複合顕微鏡には倍率を高めるためのレンズが複数あります(図1)。複合顕微鏡は、物体の画像を作成するために光を収集するオブジェクトの近くに対物レンズを持っています。これは、画像を拡大するアイピース(眼レンズ)によって拡大されます。対物レンズとアイピースを組み合わせることで、単一レンズ単一レンズを使用するよりも高い倍率を実現します。通常、複合顕微鏡は、異なる倍率(1、2)を可能にするために、様々な力の複数の対物レンズを持っています。ここでは、グラム染色、カプセル染色、内胞汚れを用いて細菌を可視化する方法について説明します。

図1:典型的な化合物顕微鏡。顕微鏡の最も重要な部分は標識される。
デンマークの細菌学者ハンス・クリスチャン・グラム(1)によって1884年に開発されたグラム染色は、細胞壁(1、2、3、4)の組成に基づいて細菌を分化する。簡単に言えば、細菌のスミアを顕微鏡スライド上に置き、細胞をスライドに付着させ、より容易に汚れを受け入れられるように熱固定する(1)。熱固定サンプルはクリスタルバイオレットで染色され、細胞を紫色に変えます。スライドは、細胞壁にクリスタルバイオレットを固定するヨウ素溶液で洗い流され、その後、非固定クリスタルバイオレットを洗い流すためにデカラーライザー(アルコール)が続きます。最後のステップでは、カウンターステイン、サフラニン、赤色色セルに添加されます(図2)。グラム陽性細菌は、デカラーライザーによって容易に浸透しない厚いペプチドギリカン層に起因する紫色に染色します。グラム陰性菌は、その薄いペプチドギリカン層と高い脂質含有量を有し、脱色剤で脱染し、サフラニンを添加すると赤色に染色される(図3)。グラム染色は、細胞を2種類(グラム陽性とグラム陰性)に分化するために使用され、細胞形状(球体または球体、ロッド、曲面ロッド、スパイラル)と配置(単一細胞、ペア、チェーン、グループ、クラスター)を区別するのにも役立ちます(1,3).
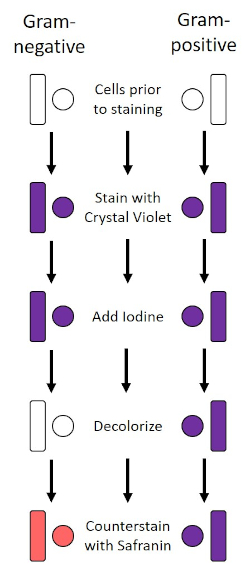
図2:グラム染色プロトコルの概略図。左の列は、プロトコルの各ステップでグラム陰性細菌がどのように反応するかを示しています。右の列は、グラム陽性の細菌がどのように反応するかを示しています。また、図示されているのは、バチル(または棒)と球体(または球)の2つの典型的な細菌細胞形状である。
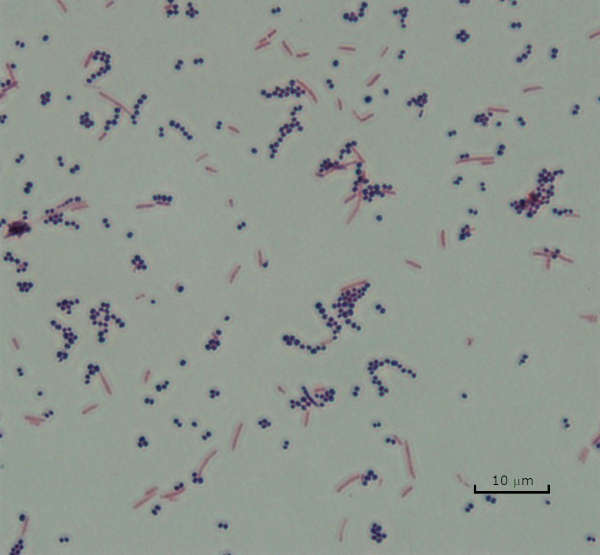
図3:グラム染色結果。黄色ブドウ球菌(グラム陽性紫色のコッカス)と大腸菌(グラム陰性赤い棒)の混合物のグラム染色。
いくつかの細菌は、カプセル(3、5)と呼ばれる細胞外粘性外層を産生する。カプセルは、表面や他の細菌への付着、乾燥からの保護、および咽頭症からの保護を含むが、これらに限定されない様々な機能を有する保護構造である。カプセルは、通常、95%以上の水を含む多糖類で構成されていますが、一部はポリアルコールとポリアミン(5)を含むことがあります。そのほとんど非イオン性組成物と汚れを撃退する傾向があるため、単純な染色方法はカプセルでは動作しません。その代わりに、カプセル染色は、細胞および背景を染色する負の染色技術を使用し、カプセルを細胞の周りに明確なハローとして残す(図4)。カプセル染色は、細菌サンプルを顕微鏡スライド上の酸性染色に塗りつぶすることを含む。グラム染色とは異なり、細菌の汚れはカプセル染色中に熱固定されません。熱固定は、カプセルを破壊または脱水することができ、偽陰性(5)につながる。さらに、熱固定は細胞を収縮させることができ、その結果、カプセルと間違えられ得る細胞の周りのクリアリングを行うことができ、偽陽性(3)を引き起こす。酸性染色は、スライドの背景を着色します。基本的な染色をフォローアップしながら、クリスタルバイオレットは、細菌細胞自体を着色し、カプセルを染色されず、細胞とスライドの背景との間に明確なハローとして現れる(図5)。従来、インドのインクは、これらの粒子がカプセルに浸透できないため、酸性染色剤として使用されてきました。したがって、カプセルも細胞もインドのインクによって染色されていない。代わりに、背景が汚されます。コンゴレッド、ニロシン、またはエオシンは、インドのインクの代わりに使用することができます。カプセル染色は、患者サンプルから培養物を見て、適切な患者治療を導くときに医師が細菌感染症を診断するのに役立ちます。封入細菌によって引き起こされる一般的な疾患には、肺炎、髄膜炎、およびサルモネラ症が含まれる。
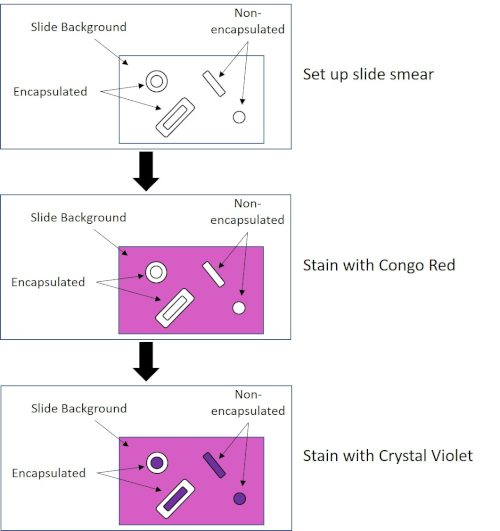
図4:カプセル染色プロトコルの概略図。上部パネルは、汚れ塗布前のスライドスミアを示しています。中央のパネルは、スライドと細菌が一次染色、コンゴレッドの後にどのように見えるかを示しています。最後のパネルは、スライドと細菌がカウンターステイン、クリスタルバイオレットの後にどのように見えるかを示しています。
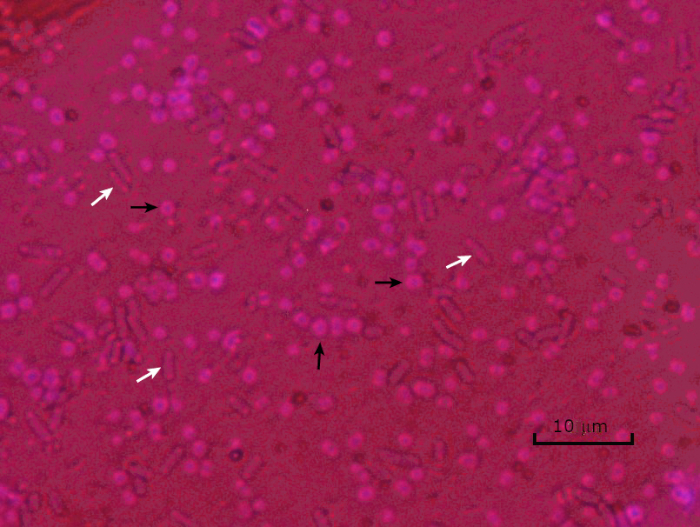
図5:カプセル染色結果。封入されたアシネトバクター・バウマンニ(黒い矢印で示される)および非封入大腸菌(白い矢印で示される)のカプセル染色。背景が暗く、A.バウマンニ細胞が紫色に染色されていることに注意してください。A.バウマンニ細胞の周りのカプセルはハローとして明らかであるが、大腸菌はハローを持たない。
有害な条件(例えば、栄養制限、極端な温度、脱水)では、一部の細菌は内在性胞子を産生し、物理的および化学的損傷に対して耐性のある代謝的に不活性な構造(1、2、8、9)を産生する。内胞子は、細菌が細胞の遺伝物質を保護することによって過酷な条件を生き残ることを可能にします。いったん条件が成長に有利な状態になったら、胞子は発芽し、細菌の増殖が続く。エンドスポールは、通常染色に使用される染料に対して不透過性であるため、標準的な染色技術では染色が困難です(1,9)。内胞子を染色するために日常的に使用される技術は、一次染色マラカイトグリーン、細胞材料に比較的弱く結合する水溶性染色、および熱を使用して、汚れを壊すことを可能にするシェーファーフルトン法(図6)です。胞子の皮質を通して(図7)。これらのステップは、成長する細胞(内胞子生物学の文脈で栄養細胞と呼ばれる)、ならびに内胞子および任意の自由胞子(もはや前の細胞の封筒内になくなっているもの)を着色する。栄養細胞は、マラカイトグリーンを除去するために水で洗浄されます。内胞子は胞子内のマラカイトグリーンを加熱することに起因する汚れを保持します。最後に、栄養細胞をサフラニンで逆染色して可視化する(図8)。エンド胞子の染色は、細菌を胞子原型および非胞子フォーマーに分化させるだけでなく、胞子がサンプル中に存在するかどうかを決定するのに役立ち、もし存在する場合、発芽時に細菌汚染を引き起こす可能性がある。
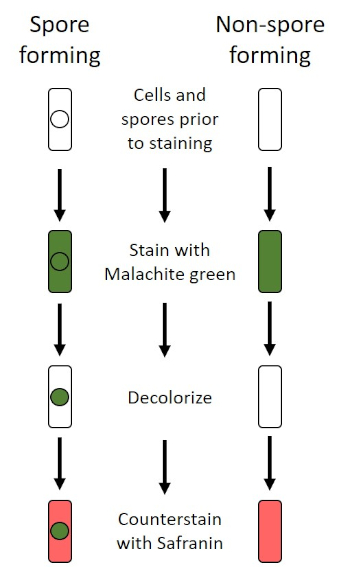
図6:内胞染色プロトコルの概略図。左の列は、胞形細菌がプロトコルの各ステップでどのように反応するかを示しています。右の列は、非胞毛形成細菌がどのように反応するかを示しています。
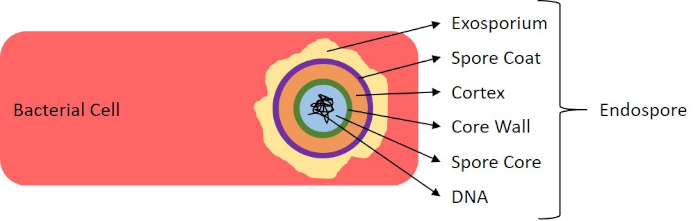
図7:内胞構造図種々の胞直構造を標識した内胞を含む細菌細胞。
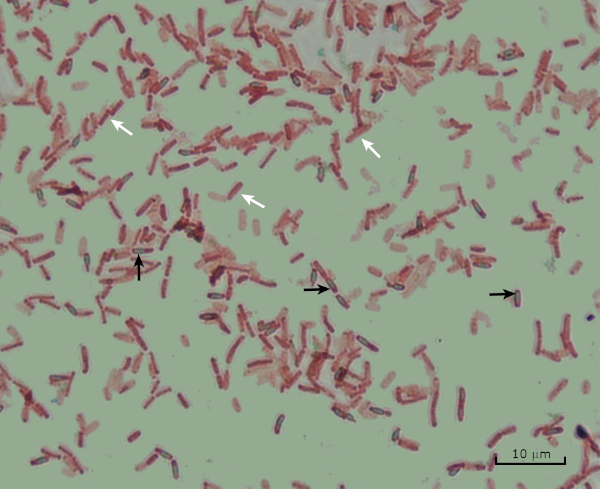
図8:内胞染色結果。バチルス・サビリスのエンドスポ胞の典型的な染色.栄養細胞(白い矢印で示される)は赤く染色され、内胞子(黒い矢印で示される)は緑色に染色されます。
Procedure
1. グラム染色
-
セットアップ
- 染料が手や衣類を汚すので、手袋と不燃性のラボコートを着用してください。
- ブンゼンバーナーは、細菌を加熱するために使用されます。炎を扱うときは注意してください。長い髪を結び返す
- 市販のグラム染色試薬が使用されます。
- 実験室のワイプが付いているきれいな顕微鏡のスライド。
-
プロトコル
- ピペ状10μLリン酸塩緩衝生理食生または培養ブロスをスライド上に置く。
- 細菌のコロニーを液体に塗りつぶし、薄い均一層を作り出す。
注:細菌がグラム染色結果に影響を与える細胞壁に変化を持つことができるので、24時間より古い培養物を使用しないでください(1、4)。 - 完全に空気乾燥スライド。
- 乾燥したら、炎(細菌側上)を通してスライドを通過させて熱修正細菌を4〜5回。
注:炎の中でスライドを長く保持したり、細菌細胞を歪めたりしないでください(1)。 - シンクの上で作業し、スライドレベルを保持し、完全に熱固定細菌をカバーするためにグラムのクリスタルバイオレットを適用し、45秒を保持することを可能にします。
- 余分なクリスタルバイオレットを斜めに保持し、スライド上の穏やかな間接的な水の流れを噴出し、それが染色された細菌の上に実行できるようにすることで、過剰なクリスタルバイオレットをすすいでください。細菌に直接水を噴出しないでください。
- スライドレベルをもう一度保持し、グラムのヨウ素溶液を塗布して染色された細菌を完全に覆い、45秒間放置する。
- 上記のステップ1.2.6のように余分なヨウ素をすすいでください。
- スライドを斜めに保持しながら、スライドにデカラーライザーを数滴追加し、流出が明らかになるまで染色された細菌の上にトリクルダウンさせます。通常、約 5 秒です。上記のステップ1.2.6のように、すぐに水ですすいでください。
注:これはプロトコルの重要なステップです。デカラーライザーが長すぎたり、十分に長くならない場合は、誤ったグラム染色(4)が生じる。 - もう一度スライドレベルを保持し、完全に細菌をカバーするためにグラムのサフラニンを適用し、45秒を保持できるようにします。
- 上記のステップ1.2.6のように過剰なサフラニンをすすいでください。
- ブロットは、ペーパータオルを使用してスライドから余分な水をこすらない。
- 100Xの目的で油浸を使用して顕微鏡上のスライドを調べます。
-
結果とデータ分析
- グラム陽性の細菌は紫色に染色します。
- グラム陰性の細菌は赤く染まる。
- 細菌の形状(コッチ、バチリ、曲面ロッド、スパイラル)が見えるようになります。
- 細菌細胞(単一細胞、対化細胞、細胞鎖、クラスター、グループ化)の配置が表示されます。
2. カプセル染色
-
セットアップ
- 手袋とラボコートを染色物として着用し、手や衣類を汚します。
- 1%のクリスタルバイオレット溶液を調作成するには、溶解するまで25mLの蒸留水で0.25グラムのクリスタルバイオレットを混ぜます。
- 1%のコンゴ赤溶液を調作成するには、0.25グラムのコンゴレッドを溶かすまで25 mLの蒸留水と混合します。
- 実験室のワイプが付いているきれいなスライド。
-
プロトコル
- スライド上に10 μLコンゴレッドを配置します。
- ピペット先端を使用して、細菌のコロニーを染料に塗りつぶし、薄い均一層を作り出します。
- 染料/細胞混合物と完全に空気乾燥スライド、5-7分。
注:加熱はカプセルを脱水または歪めることができるので、加熱固定しないでください。 - 1%クリスタルバイオレットで1分間塗りつぶしを水浸しにします。
- スライドを斜めに保持し、滑り台に穏やかな間接的な水の流れを吹き付け、染色された細菌の上に流れ落ちることによって余分な汚れをすすいでください。細菌に直接水を噴出しないでください。
- 完全に空気乾燥するまで、スライドを45度の角度で保持します。
- 100Xの目的で油浸の下で顕微鏡の汚れを調べる。
-
結果とデータ分析
- 細菌細胞は紫色に染色します。
- スライドの背景が暗く染まる。
- カプセルは、暗い背景に対して細胞の周りの明確なハローになります。
3. 内散性染色(シェーファーフルトン法)
-
セットアップ
- 手袋と不燃性のラボコートを着用して、手や衣類を染料や炎から守ってください。
- ブンゼンバーナーは、細菌を加熱するために使用されます。炎を扱うときは注意してください。長い髪を結び返す
- 0.5%のマラカイトグリーン溶液を調作成するには、溶解するまで25 mLの蒸留水で0.125グラムのマラカイトグリーンを混ぜます。
- 市販のグラムのサフラニン試薬ソリューションを使用してください。
- 実験室のワイプが付いているきれいなスライド。
-
プロトコル
- ピペ状10μlリン酸緩衝生理食生(PBS)または培養ブロスをスライド上に置く。
- 無菌技術を用いて、細菌のコロニーを液体に塗りつぶし、薄い偶数層を作り出す。
注:内胞子は一般に若い細胞では形成されないため、培養は18~36時間齢(9)であることが推奨される。 - 完全に空気乾燥スライド。
- 炎を4~5回通してスライド(細菌側を上に)渡すことで熱を固定します。
- 染料を含むのに役立つように、熱固定スミアの上にレンズペーパー(細菌の汚れに合わせてカット)を置きます。
- マラカイトグリーン溶液でレンズペーパーを飽和させます。
- 熱いお湯のビーカーの上にスライドを置き、5分間スチームスライドし、必要に応じて一度にさらに染料を加えてレンズペーパーを湿らせます。
注:染料溶液の過熱や乾燥は避けてください。 - ビーカーからスライドを取り外し、レンズペーパーを取り外して捨て、スライドを2分間冷まします。
- スライドを斜めに持ち、穏やかな間接的な水の流れをスライドに吹き付け、スミアの上に排水できるようにし、十分に洗い流します。
- スライドレベルを保持し、サフラニンで洪水スミアは、1分間立つことを可能にする。
- 上記のステップ 3.2.9 のように過剰なサフラニンをすすいでください。
- 空気乾燥を許可します。
- 100Xの目的で油浸の下の顕微鏡の滑土を調べる。
-
結果とデータ分析
- 胞子は緑色に染まる
- 栄養細胞は赤く染まる。
- いくつかの栄養細胞は胞子を含む;細胞は赤く染色し、内胞子は緑色に染まる。
細菌は、形状、細胞の配置、カプセルを産生するかどうか、胞子を形成するかどうかなど、多くの特徴を持つ顕微鏡生物です。これらの特徴はすべて染色によって視覚化され、異なった細菌種の同定そして分類を助けることができる。
細胞の形状と配置の最初の2つの特徴を調べるために、グラム染色と呼ばれる簡単な技術を使用することができます。ここで、結晶バイオレットは、スライド上に熱固定された細菌に適用される。その後、デカラーライザーが適用され、厚いペプチドギリカン層を持つ細菌は、この層がデカラーライザーによって容易に浸透しないので、紫色に染色します。これらの細菌はグラム陽性と呼ばれる。
グラム陰性細菌は、より薄いペプチドギリカン層を持っており、紫色を失い、脱色剤を脱汚します。しかし、サフラニンカウンター染色を加えると赤みがかったピンク色に染まり、外側のリポ多糖層に結合します。一度染色されると、細胞は、鎖やクラスターなどの形態、大きさ、および配置のために観察することができ、分類および同定にさらに役立ちます。
微生物学者のツールキットのもう一つの有用な技術は、いくつかのタイプの細菌細胞を取り囲む外部カプセルを視覚化するために使用されるカプセル染色です。カプセル非イオン組成物と汚れを撃退する傾向があるため、単純な染色方法は機能しません。代わりに、細菌細胞が結晶紫色で染色される前に、まずコンゴ赤などの酸性着色剤で背景を染色する負の染色技術が使用されます。これは、細胞の周りに明確なハローとして存在する任意のカプセルを残します。
ここでカバーされる最終的な主要な染色技術は、研究されている細菌が胞子を形成するかどうかを判断するのに役立ちます。悪条件では、一部の細菌は、極端な温度や脱水のような環境ストレスの期間を通じて細菌の生存を確保することである内胞子、休眠、タフ、非生殖構造を生成します。しかし、すべての細菌種が内生胞を作るわけではないため、多くの染料に対して不浸透性であるため、標準的な技術で染色することは困難です。シェーファーフルトン法は、スライドに固定された細菌に適用されるマラカイト緑色の汚れを使用しています。スライドは、サフラニンで染色される前に水で洗浄されます。栄養細胞はピンクがかった赤色に見え、存在する内胞子は緑色で表示されます。このビデオでは、これらの一般的な細菌染色技術を実行し、光顕微鏡を使用して染色サンプルを調べる方法を学びます。
手順を開始するには、長い髪を結び、ラボコートや手袋を含む適切な個人用保護具を着用してください。
その後、実験室の拭き取りで新鮮な顕微鏡スライドをきれいにします。次に、1Xリン酸のピペット10マイクロリットルを第1スライド上に緩衝生理食塩水を入れた。次に、滅菌ピペット先端を使用して、LB寒天プレートから単一の細菌コロニーを選択する。液体中の細菌コロニーを塗りつぶし、薄く、均一な層を作り出す。ベンチ上部にスライドをセットし、完全に空気乾燥させます。
乾燥したら、ブンゼンバーナーを点灯させて細菌を加熱します。トングを使用して、バーナー炎を数回通り、細菌の側面を上にして、あまりにも長く炎の中でスライドを保持しないように注意して、細胞を歪める可能性があります。
さて、シンクの上で作業し、スライドレベルを保持し、完全に細菌の汚れをカバーするためにグラムのクリスタルバイオレットの数滴を適用し、その後、45秒間立つためにベンチにスライドを置きます。次に、スライドを斜めに保持し、スライドの上部に水の流れをそっと噴き出し、細菌の汚れを直接噴出しないように注意してください。さて、もう一度スライドレベルを保持し、グラムのヨウ素溶液を塗布して染色された細菌を完全に覆い、さらに45秒間放置します。次に、前に示したように、スライドからヨウ素を慎重にすすいでください。スライドを斜めに保持したまま、Gramのデカラー化剤をスライドに数滴加え、染色された細菌の上を約5秒間実行できるようにします。直ちに、前に示したように水ですすいでください。これにより、スミアの過色化が制限されます。次に、スライドレベルをもう一度保持し、グラムのサフラニンカウンターステインを塗布し、染色された細菌を完全に覆います。45秒後、スライドからサフラニンを水で軽くすすいでから、ペーパータオルで乾かします。
最後に、浸漬油の滴をスライドに直接追加し、その後、100Xオイル対物レンズで軽微顕微鏡を使用してスライドを調べます。
この染色プロトコルを開始するには、まず正しい個人用保護具を着用し、次に使用するガラススライドがきれいであることを確認します。
次に、ソリューションを準備します。1%の結晶バイオレット溶液を作るために、0.25グラムの結晶紫色粉末を25ミリリットルの蒸留水と混合し、溶解するまで渦を混ぜます。次いで、0.25グラムのコンゴ赤色粉末を25ミリリットルの蒸留水と渦を溶かして溶かして1%コンゴ赤溶液を調製する。さて、スライド上のコンゴ赤溶液のピペット10マイクロリットル。クリーンな無菌ピペットチップを使用して、LB寒天プレートから単一の細菌コロニーを選択します。次に、細菌コロニーを染料に塗り込み、薄く均一な層を作り出す。細菌スライドを5~7分間完全に空気乾燥させます。スライドが乾燥したら、スミアをカバーするのに十分な1%の結晶バイオレットでスミアを洪水し、1分間座らせます。さて、スライドを斜めに持ち、スライドの上に水の流れをそっと噴き出し、細菌を直接噴出しないように注意してください。完全に空気が乾燥するまで、スライドを45度の角度で保持し続けます。最後に、浸漬油の滴をスライドに直接追加し、次に、100Xオイル目的の小顕微鏡を使用してスライドを調べます。
内胞スポア染色を行うには、まず、0を混合して0.5%のマラカイトグリーン溶液を調出す。125グラムのマラカイトグリーンパウダーを25ミリリットルの蒸留水で、溶解するまで溶液を渦にする。次に、スライドの中心に1X PBSのピペット10マイクロリットル。次に、滅菌ピペット先端を使用して、LB寒天プレートから単一の細菌コロニーを選択する。細菌を液体に塗りつぶし、薄くても均一な層を作り出します。次に、ベンチ上部にスライドをセットし、完全に空気乾燥させます。乾燥したら、ブンゼンバーナーを点灯させて細菌を加熱します。青いバーナーの炎を数回通り、細菌側を上に向けて渡します。次に、スライドが冷却されたら、熱固定スミアの上にプリカットレンズペーパーを置きます。次に、最も高い設定にホットプレートをオンにし、沸騰に水のビーカーをもたらします。
レンズペーパーをマラカイトグリーン溶液で飽和させ、トングを使用して、沸騰した水のビーカーの上にスライドを置き、5分間蒸します。必要に応じて、一滴ずつ多くの染料を加えることで、レンズ紙を湿らせておきます。次に、もう一度トングを使用して、ビーカーからスライドを拾い、レンズペーパーを取り外して捨てます。スライドが 2 分間冷やされます。シンクの上で作業し、スライドを斜めに保持し、スライドの上部に水の流れをそっと噴き出します。次に、スライドレベルを保持し、スライドを完全にカバーするためにサフラニンを適用します。その後、1分間放置します。次に、スライドを斜めに保持し、前に示したようにすすいで下げます。スライドをベンチ上部の空気乾燥させます。最後に、浸漬油の滴をスライドに直接追加し、その後、100Xオイルの目的で、軽い顕微鏡でスライドを調べます。
グラム染色プロトコルでは、2つの異なる色の汚れが生じる可能性があります。濃い紫色の染色は、細菌がグラム陽性であり、結晶紫色の染色を保持していることを示します。対照的に、赤みがかったピンクの染色はグラム陰性細菌の特徴であり、代わりにサフラニンカウンター染色によって着色されます。さらに、異なる形状や細菌の配置は、グラム染色後に視覚化することができます。例えば、コッチ、または丸い細菌を棒状のバチルスから区別したり、鎖を形成する細菌を同定したり、典型的に塊として凝集するものと比較して、または一体的に発生する細菌を同定することができる。
カプセル染色顕微鏡画像では、細菌細胞は通常紫色に染色され、スライドの背景は暗く染色されるべきである。この暗い背景に対して、細菌のカプセルが存在する場合、細胞の周りに明確なハローとして現れる。
最後に、内胞スポア染色では、栄養細胞はサフラニンカウンター染色によって赤く染色されます。内胞子がサンプル中に存在する場合、これらはマラカイト緑色の汚れを保持し、青色緑色に見えます。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
細菌は、その同定に役立つことができる特徴を持っています。これらの特性のいくつかは、染色および光顕微鏡検査によって観察することができる。これらの特性を観察するのに有用な3つの染色技術は、グラム染色、カプセル染色、およびエンドスポア染色である。各技術は、細菌の異なる特性を識別し、医師が患者のための治療を推奨し、サンプルや食品中の潜在的な汚染物質を特定し、サンプルの無菌性を確認するのに役立ちます。
Subscription Required. Please recommend JoVE to your librarian.
References
- Black, J. G. Microbiology Principles and Explorations, 4th edition. Prentice-Hall, Inc., Upper Saddle River, New Jersey. (1999)
- Madigan, M. T. and J. M. Martinko. Brock Biology of Microorganisms, 11th edition. Pearson Prentice Hall, Upper Saddle River, New Jersey. (2006).
- Leboffe, M. J., and B. E. Pierce. A Photographic Atlas for the Microbiology Laboratory, 2nd ed. Morton Publishing Company, Englewood, Colorado. (1996).
- Smith, A. C. and M. A. Hussey. Gram stain protocols. Laboratory Protocols. American Society for Microbiology, Washington, DC. Available from: http://www.asmscience.org/content/education/protocol/protocol.2886. (2005).
- Hughes, R. B. and A. C. Smith. Capsule Stain Protocols Laboratory Protocols. American Society for Microbiology, Washington, DC. Available from: http://www.asmscience.org/content/education/protocol/protocol.3041. (2007).
- Anthony, E. E. Jr. A note on capsule staining. Science 73(1890):319-320 (1931).
- Finegold, S. M., W. J. Martin, and E. G. Scott. Bailey and Scott's Diagnostic Microbiology, 5th edition. The C. V. Mosby Company, St. Louis, Missouri. (1978).
- Gerhardt, P., R. G. E. Murray, W. A. Wood, and N. R. Krieg. Methods for general and molecular bacteriology. ASM Press, Washington, DC. (1994).
- Hussey, M. A. and A. Zayaitz. Endospore Stain Protocol. Laboratory Protocols. American Society for Microbiology, Washington, DC. Available from: http://www.asmscience.org/content/education/protocol/protocol.3112. (2007).