Once the contractility and transient tracings are collected (Figure 2), data are easily analyzed with the appropriate software. The whole tracing, or portions thereof can be averaged (Figure 3). Contractility can be analyzed in a variety of ways. For systolic function, one can assess the magnitude of contraction with fractional shortening, or speed of contraction with time to peak shortening, and contraction velocity. Diastolic function can be analyzed similarly with time to 50% shortening and relaxation velocity. For calcium transients, similar data can be compared including baseline and peak Fura-2 ratios and time to peak or baseline (Table 8). These analyses provide comparable data from WT and KO hearts on systolic and diastolic cellular contractile function, as well as calcium flux, any of which could be altered in a heart with similar whole organ dysfunction. Additionally, cells from the same isolation can be used for other experiments as listed in the protocol, including immunohistochemical staining (Figure 4).
| Ca2+ free Tyrode | mM | FW/concentration | Amount for 2 L solution |
| NaCl | 135 | 58.44 g | 15.8 g |
| KCl | 4 | 74.56 g | 0.596 g |
| MgCl2 | 1 | 1 M | 2 ml |
| HEPES | 10 | 238.31 g | 4.77 g |
| NaH2PO4 | 0.33 | 141.96 g | 0.094 g |
Table 1. Calcium-free Tyrode’s solution – Reagents for 2 L of stock Ca2+-free Tyrode’s solution.
| 1.2 mM Ca2+ Tyrode | mM | FW/concentration | Amount for 2 L solution |
| NaCl | 137 | 58.44 g | 16 g |
| KCl | 5.4 | 74.56 g | 0.805 g |
| MgCl2 | 0.5 | 1 M | 1 ml |
| HEPES | 10 | 238.31 g | 4.77 g |
| CaCl2·2H2O | 1.2 | 147.01 g | 0.353 g |
Table 2. 1.2 mM Calcium Tyrode’s solution – Reagents for 2 L of stock 1.2 mM Ca2+ solution.
| Perfusion Buffer | mM | FW | Amount in 150 ml Ca2+ free Tyrode |
| Glucose | 10 | 180.16 g | 0.27 g |
| 2,3-Butanedione-monoxime (BDM) | 10 | 101.11 g | 0.152 g |
| Taurine | 5 | 125.16 g | 0.094 g |
Table 3. Perfusion Buffer – Reagents to prepare perfusion buffer on the day of experiment.
| Buffer A | 35 ml Perfusion Buffer |
| Bovine serum albumin (BSA) | 0.175 g |
Table 4. Buffer A – Reagents to prepare Buffer A on the day of experiment.
| Buffer B | mM | FW | 25 ml Ca2+ Tyrode buffer |
| Glucose | 5 | 180.16 g | 0.0225 g |
Table 5. Buffer B – Reagents to prepare Buffer B on the day of experiment.
| Transfer Buffer | Buffer A (ml) | Buffer B (ml) |
| 0.06 mM Ca2+ | 9.5 | 0.5 |
| 0.24 mM Ca2+ | 8 | 2 |
| 0.6 mM Ca2+ | 5 | 5 |
| 1.2 mM Ca2+ | 0 | 10 |
Table 6. Transfer Buffer – Reagents to prepare transfer buffer on the day of experiment.
| Enzyme Digestion Buffer | 25 ml Perfusion Buffer |
| Collagenase B | 0.4 mg/g body weight |
| Collagenase D | 0.3 mg/g body weight |
| Protease XIV | 0.05 mg/g body weight |
Table 7. Enzyme Digestion Buffer – Reagents to prepare enzyme digestion buffer on the day of experiment.
| Contractility | |
| Baseline | 1.73 μm |
| Peak | 1.64 μm |
| Fractional Shortening | 4.70% |
| Time to peak | 0.056 sec |
| Time to 50% baseline | 0.043 sec |
| Calcium transients | |
| Baseline | 1.18 |
| Peak | 1.32 |
| Time to peak | 0.021 sec |
| Time to 50% baseline | 0.083 sec |
Table 8. Contractility and calcium transient analyses. N = 5.
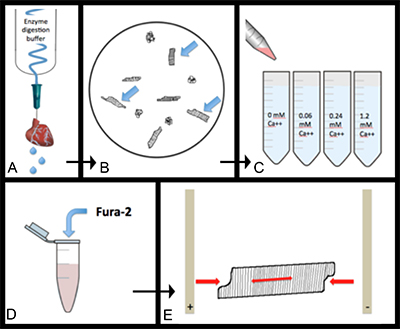
Figure 1. General overview of experimental design. A) Heart is digested via coronary perfusion with enzyme digestion buffer. B) Cells are further dispersed into individual, healthy myocytes. C) Cells are sequentially transferred through increasing calcium concentrations so cells become calcium tolerant. D) Cells are loaded with the calcium dependent dye, Fura-2 AM. E) Loaded cells are electrically paced while sarcomere length, cell length, and fluorescence are recorded. Please click here to view a larger version of this figure.
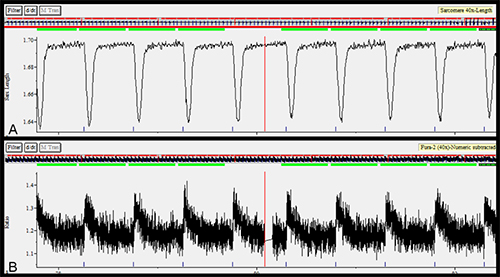
Figure 2. A) Sample contractility tracing represented by sarcomere length vs. time. B) Sample calcium transients represented by Fura-2 ratio vs. time. Please click here to view a larger version of this figure.
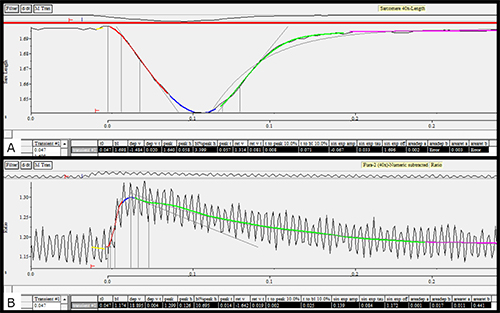
Figure 3. A) Average contractility tracing after analysis. B) Average of calcium transients after analysis. Please click here to view a larger version of this figure.

Figure 4. Immunofluorescent staining of cardiomyocyte cytoskeleton (α-actinin).
