With successful staining, TUNEL-positive signal should be bright enough to isolate from autofluorescence by setting intensity thresholds. TUNEL-positive objects at low magnification may appear as bright irregular fragments in skeletal muscle (Figure 1A). However, at higher magnification, some TUNEL-positive objects with the classic apoptotic morphology should be observed, if the cell death type involved is apoptosis (Figure 1B). The positive control (DNase added) should exhibit abundant TUNEL-positive signal, distributed uniformly across all tissues in the section (Figure 1C). The negative control (TdT enzyme omitted from reaction) should yield low-intensity background and autofluorescent signal only (Figure 1D). Skin provides an internal positive control in whole leg sections, as this tissue has a high rate of normal apoptosis (Figure 1A).
Red blood cells, bone, and endothelial cells may contribute significant autofluorescent signal (Figure 2). It is advisable to image each section in a separate channel with no fluorescent labeling to ensure that autofluorescence is not mistaken for positive TUNEL signaling. True TUNEL-positive signal should have much higher intensity than autofluorescence, so that it can be isolated by setting image intensity thresholds (Figure 2A, middle row). Alternatively, the autofluorescent channel may be digitally subtracted from the TUNEL channel to yield TUNEL-positive signal (Figure 2A, bottom row).
The TUNEL labeling method described here has been used to quantify skeletal muscle cell death in a mouse model of SMA10. TUNEL labeling shows an increase in the number of apoptotic profiles in leg muscles from 5 day old SMA mice, compared with littermate controls (Figure 3A). The increase in apoptosis is quantifiable by the method described above, indicating statistically significant differences in multiple muscle groups (Figure 3B).
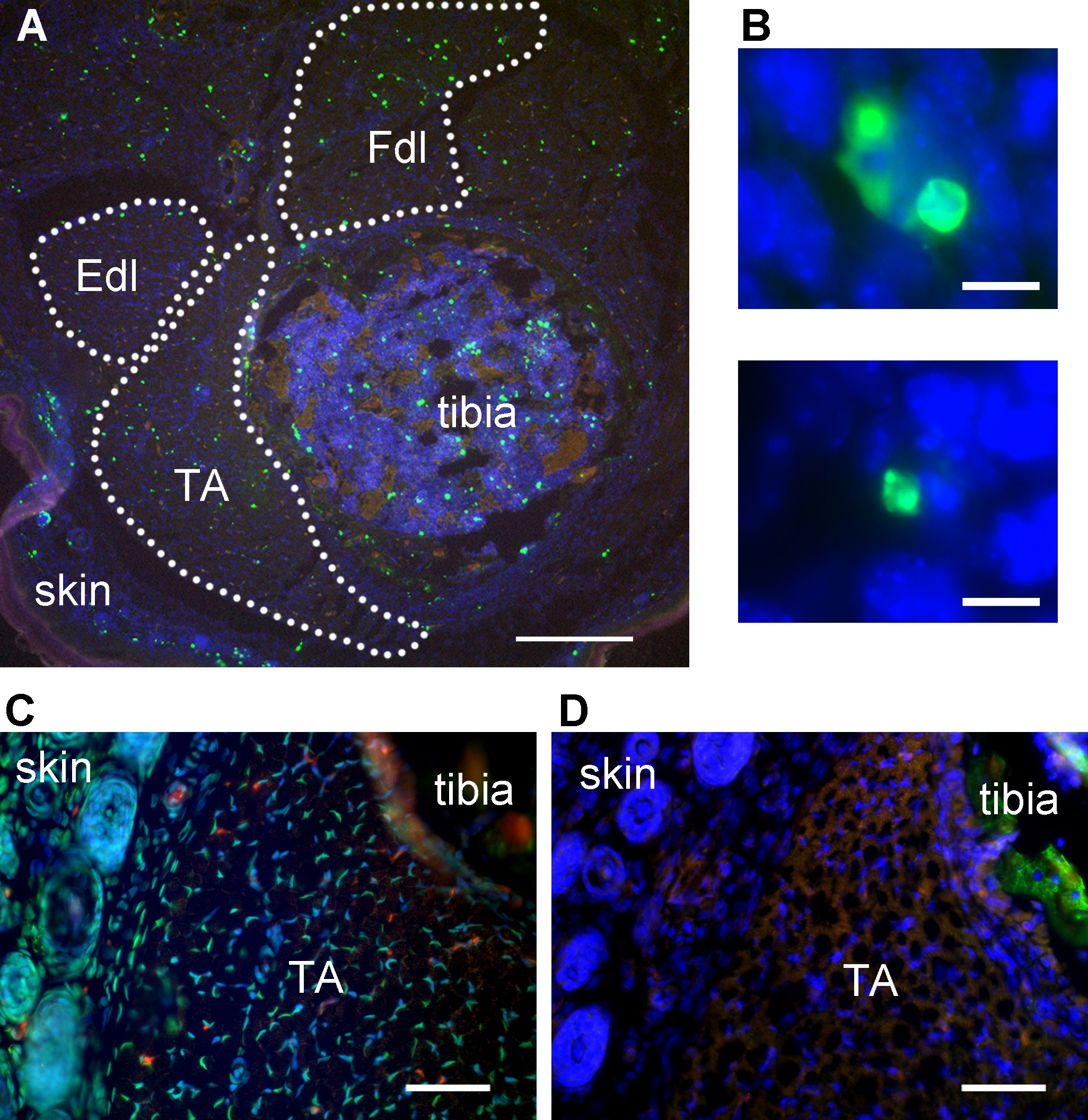
Figure 1: Representative TUNEL labeling of mouse hindlimb muscles. (A) TUNEL-labeled transverse section of mouse hindlimb showing tibialis anterior muscle, tibia, and skin. TA – tibialis anterior, Fdl – flexor digitorum longus, Edl – extensor digitorum longus. Green – TUNEL, blue – nuclei, red – autofluorescent signal. Dotted lines delineate muscle areas for quantitation. Skin adjoining hindlimb muscles provides an internal positive control for TUNEL labeling. Scale bar = 200 μm. (B) Representative high-magnification images identifying TUNEL-positive structures (green) in embryonic day 13 mouse skeletal muscle as apoptotic nuclei. Scale bar = 10 μm. A and B modified from Fayzullina and Martin 201410. (C) Transverse section of mouse hindlimb TUNEL-labeled after DNase treatment to serve as a positive control. Most nuclei have bright TUNEL labeling. Scale bar = 50 μm. (D) Transverse section of the same mouse hindlimb as shown in C (semi-adjacent to section in C) TUNEL-labeled with TdT enzyme omitted to serve as a negative control. There is no bright TUNEL-positive signal; the only green signal is autofluorescence as detected by colocalization with signal in the red channel. Scale bar = 50 μm. Please click here to view a larger version of this figure.
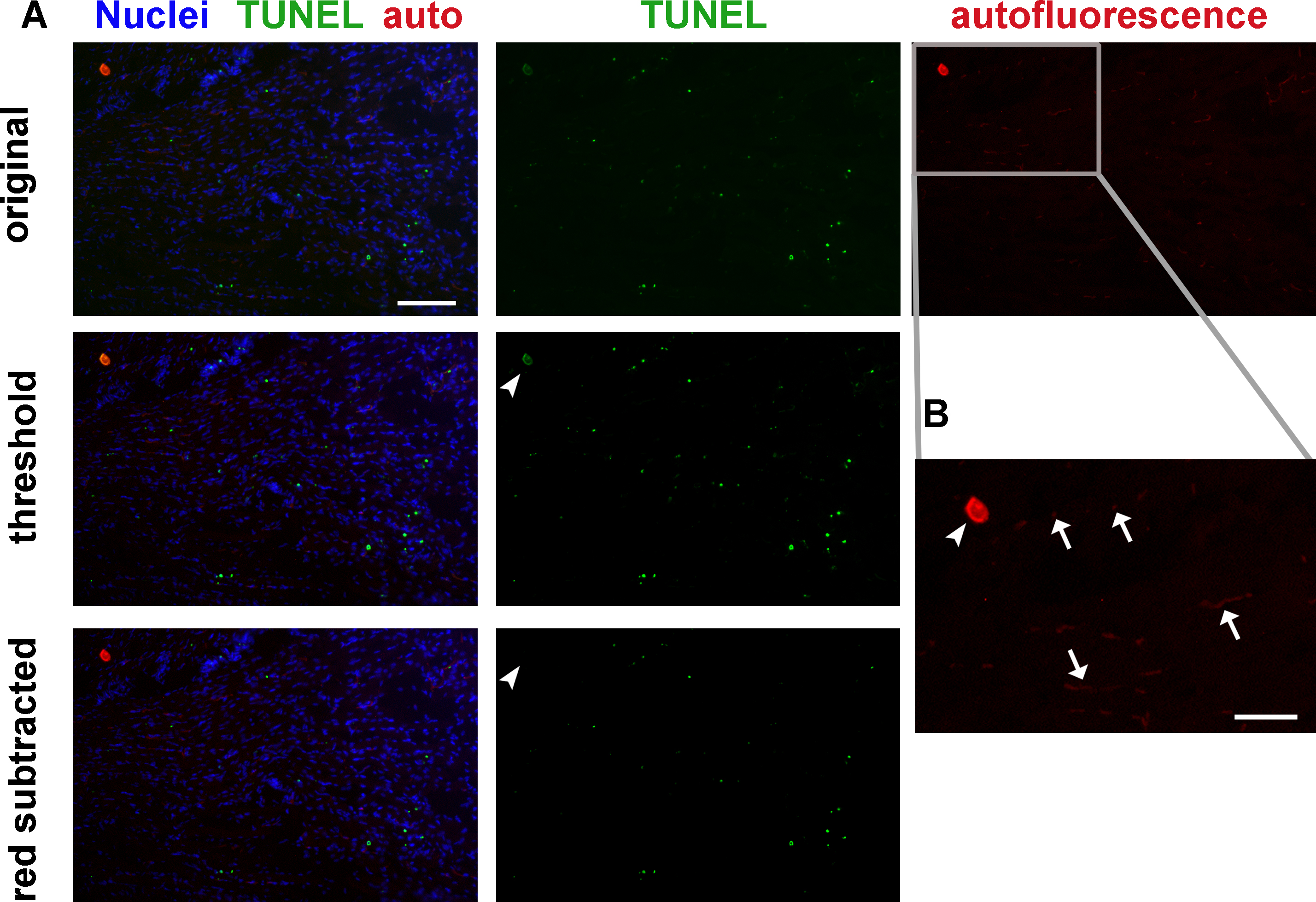
Figure 2: Tissue autofluorescence must be considered and excluded from quantitation when analyzing TUNEL labeling. (A) Mouse hindlimb transverse section with TUNEL labeling and autofluorescence: original image (top row), same image with adjusted thresholds (middle row), and same image with autofluorescence (red) channel subtracted (bottom row). Thresholding eliminates most red blood cell and endothelial cell autofluorescence but not the staining artifact (arrowhead, middle row). Channel subtraction also eliminates the staining artifact (arrowhead, bottom row). Green – TUNEL, red – autofluorescence, blue – nuclei. Scale bar = 100 μm. Gray rectangle delineates area magnified in B. (B) Higher magnification image of red autofluorescence panel in A. Red blood cells and some endothelial cells (arrows), and a tissue staining artifact (arrowhead) autofluoresce in both red and green fluorescence filters. Scale bar = 50 μm. Please click here to view a larger version of this figure.

Figure 3: TUNEL labeling shows cell death in the skeletal muscle of neonatal SMA mice. Lower hindlimb muscles from SMA mice at postnatal day 5 were labeled by the TUNEL method. (A) TUNEL labeling shows multiple apoptotic profiles in multiple muscle groups in SMA mouse muscle (right) compared with littermate control (left). Green – TUNEL, blue – Hoechst (nuclei), red – autofluorescence. Dotted lines delineate the muscle areas quantified. TA – tibialis anterior, Fdl – flexor digitorum longus, Edl – extensor digitorum longus. Scale bars = 200 μm. (B) TUNEL labeling was quantified as total pixel area of TUNEL signal normalized to total pixel area of muscle. Means ± standard errors are shown, n = 4 – 5 mice. Statistical significance between SMA and control for each muscle group (one-tailed t-test): * p <0.05, ** p <0.01, *** p <0.001. This figure has been modified from Fayzullina and Martin 201410. Please click here to view a larger version of this figure.
