As a validation of this approach we performed a representative chemical genetic interaction screen of the chemotherapeutic agent 5-fluorouracil (5-FU) following the above outlined protocol. 5-FU is known to disrupt thymidylate synthase as well as DNA and RNA metabolism18. The chemical genetic interactions of 5-FU are well studied and have been investigated by yeast barcode microarray techniques using both heterozygous and homozygous deletion collections8,19. Here we show that similar results can be obtained by comparative quantitation of mutant colony size.
It should be noted that the screen performed utilizes the non-essential haploid gene deletion collection. This type of screen can also be performed using diploid strains, temperature-sensitive mutants, and hypomorphic alleles, allowing the inclusion of essential gene mutants. One advantage to utilizing the haploid deletion collection is increased drug sensitivity of target pathways, given the complete absence of the gene product. However, two significant disadvantages are that essential gene hypersensitivity cannot be queried, and the haploid collection is prone to spontaneous suppressor mutations that are selected for over multiple propagations. This can lead to deterioration of the collection. Thus, there is an inherent trade off between the integrity and breadth of the array and sensitivity to drug effect. This must be taken into consideration and the most appropriate mutant collection selected based on the desired application.
First, the appropriate sub-lethal concentration of 5-FU to be used in the screen was determined to be 10 µM by growing BY4741 (MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0) cells on increasing concentration of 5-FU and visually evaluating yeast colony growth (Figure 3A). Alternatively, similar results can be obtained by growing yeast in microtiter plates in liquid culture and frequent monitoring of the cultures optical density using a microplate reader (Figure 3B), as outlined by previous groups10,20.
Using the Singer ROTOR, the yeast DMA was replicated at 1,536 colonies per plate density onto media containing 10 µM 5-FU or a dimethyl sulfoxide (DMSO) control (Figure 4A). These plates were incubated for 24 hr at room temperature and imaged on a flat bed scanner. Colony size measurement for each mutant on both experimental and control plates was performed using the Balony image analysis engine15. The Balony software calculates the size ratio of colonies on the experimental array relative to control. The user is able to set a ratio threshold and if the screen is performed in at least triplicate a p-value will be assigned for each mutant. In our representative screen mutants that showed a ratio of ≤0.8 were considered to be hits. The results of the colony quantification can be presented graphically by plotting the size ratio for each colony on the y-axis and the gene deletion ordered by array position (Figure 4B) or ratio (Figure 4C) on the x-axis.
Synthetic sick/lethal mutants identified in the screen can be grouped based on functional descriptions by performing gene ontology (GO) analysis. There are several publically available tools for GO analysis of lists of S. cerevisiae genes; including Funspec21, DAVID Bioinformatics Database22,23, and the Saccharomyces Genome Database (SGD) Go Term Finder24. Gene deletions that had a ratio of less than or equal to 0.8 in the 5-FU screen were used as an input list for Funspec. Funspec accepts a list of systematic or common yeast gene names and outputs summaries of gene ontologies, functional classifications, localization, protein complexes as well as other useful classifications that are enriched in that list. The representative screen performed showed an enrichment of ontologies for RNA metabolism, including tRNA wobbles, uridine modification and RNA surveillance machinery, and response to DNA damage (Table 1). These results agree with previous studies that have shown 5-FU treatment results in the accumulation of polyadenylated noncoding RNAs, which are processed by the nuclear Trf/Rrp6/Air/Mtr4 Polyadenylation (TRAMP) exosome25. Thus these findings are in agreement with previous chemical genetic screens and the known mechanism of 5-FU bioactivity8,19.
As with all high-throughput functional genomic approaches it is necessary to validate the results. To validate chemical genetic interactions yeast mutants should first be PCR-validated with primers that anneal within the target gene’s promoter and KANMX disruption cassette. Selection of strains for validation is somewhat arbitrary, however prioritizing mutants that show a strong phenotype and group functionally provides a good starting point. Next, hypersensitivity to a chemical is confirmed using spotting assays, a common approach for measuring fitness of yeast mutants. To this end, serial dilutions of yeast strains are spotted in a grid on media containing the appropriate chemical dose as well as a control. As an example, we confirm the chemical genetic interaction of 5-FU with air1 and trf5 mutants of the TRAMP complex (Figure 5). These genes encode components of the TRAMP nuclear exosome. We note that the rrp6 strain, lacking the core 3'-5' exonuclease activity of TRAMP, was lost in our lab’s high-density DMA collection. On obtaining a fresh rrp6 mutant we confirm that rrp6 and 5-FU also display a chemical genetic interaction, establishing that TRAMP activity is required to tolerate 5-FU. This illustrates that the integrity of large deletion collections may become compromised over time, making proper DMA maintenance and strain validation critical.
Our screen also identified mutants in several DNA repair enzymes (including rad50, rad52 and mre11) as 5-FU sensitive. Scoring of colony size indicated the relative fitness of these mutants on 10 µM 5-FU as 0.7, 0.65, and 0.42 compared to wild type. Based on our confirmatory spotting assays (Figure 5), these values are an underestimate likely due to the limited dynamic range of fitness measurement by colony size scoring. This highlights a second reason to independently confirm interactions by serial spotting: the magnitude of some chemical genetic interactions can be underestimated in primary data analysis. Our results illustrate that a growth-based chemical genetic screen performed with ~4,800-strain haploid deletion collection, in triplicate, provides adequate resolution to confidently isolate specific chemical genetic interactions.

Figure 1: Schematic Representation of Chemical Genetic Interactions. Gene deletions that result in hypersensitivity to chemical perturbation allow for the identification of biological processes targeted by a chemical. (A) Convergent pathways can be disrupted by either gene deletion (geneA) or chemically (geneB). Individually, these cellular insults can be tolerated due to the inherent redundancy in biological pathways. However, in combination cell viability is compromised resulting in either a synthetically sick or lethal phenotype. (B) Deletion of genes (geneC) critical for mitigating chemically induced stress also cause hypersensitivity and indicate biological pathways disrupted by chemical treatment. Please click here to view a larger version of this figure.

Figure 2: Workflow for Chemical Genetic Screen. (A) Determination of sub-inhibitory dose: Parental yeast strains are grown to stationary phase, diluted 1:5,000, and spread plated on solid YEPD media containing increasing chemical dose. The highest concentration having no greater than 10-15% growth inhibition is selected for screening against the DMA. (B) Systematic Chemical Genetic Screen: Using a high-throughput array pinning robot the S. cerevisiae DMA is replica plated onto media containing the appropriate concentration of test chemical. Plates are incubated at room temperature for 24-48 hr, imaged, and relative colony size determined. Please click here to view a larger version of this figure.

Figure 3:. Determination of Sub-Inhibitory Concentration. (A) Growth of BY4741 cells on solid media containing 5-fluouracil. Saturated BY4741 culture diluted 1:5,000 and plated on increasing concentrations of 5-fluorouracil. (B) Growth curve of BY4741 cells in liquid media containing 5-flurouracil. ~4 × 104 cells were deposited in triplicate in increasing concentrations of 5-FU and ODs were measured every 10 min for 22 hr.
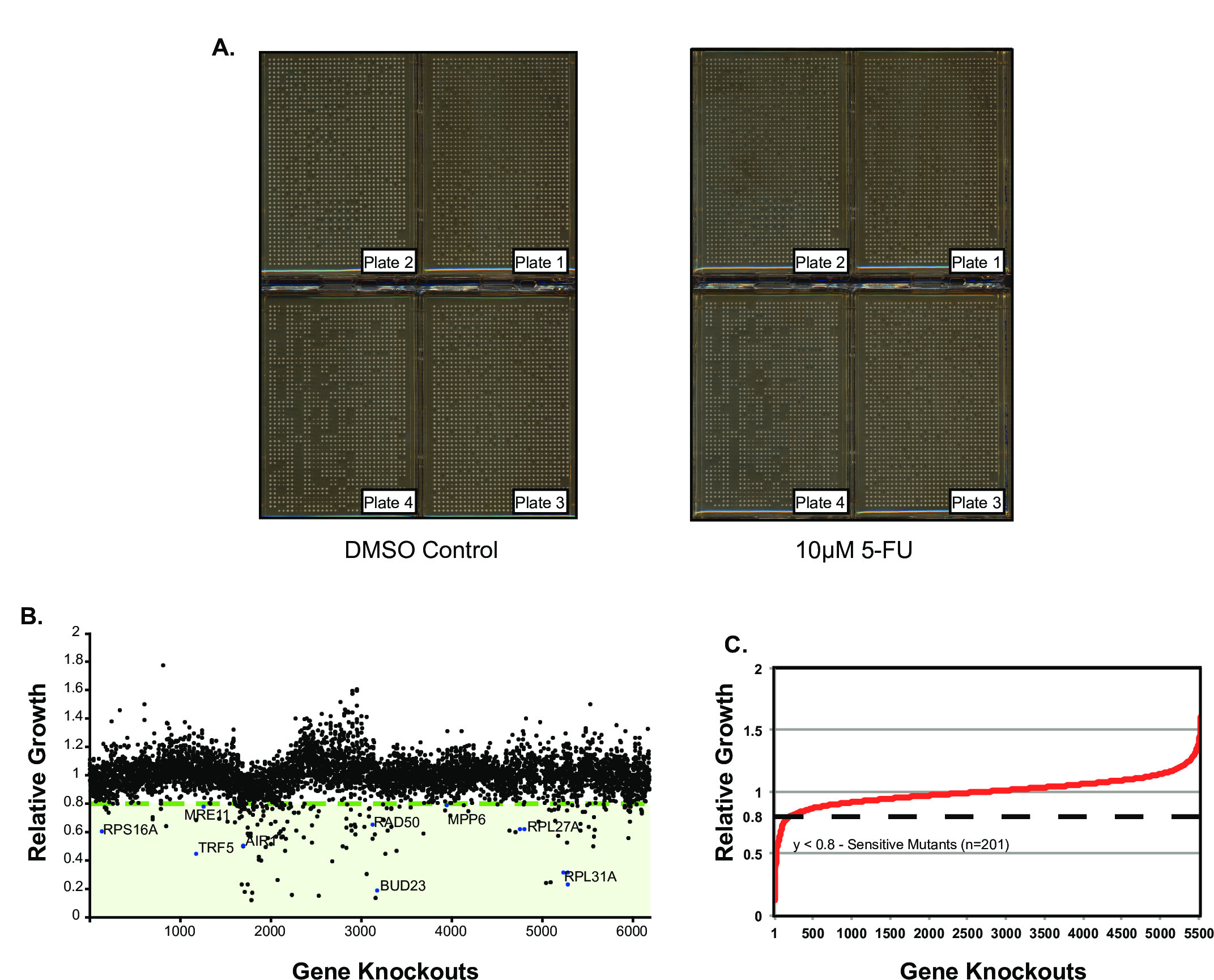
Figure 4: Chemical Genetic Screen of 5-Fluorouracil. (A) S. cerevisiae deletion mutant array replicated on YEPD agar containing 10 µM 5-FU (right) and DMSO control (left). (B) Results of Chemical Genetic Screen: Relative growth of mutants on 10 µM 5-FU compared to DMSO control ordered by array position. (C) Relative growth of mutants on 10 µM 5-FU compared to DMSO control ordered by colony size ratio. A ratio threshold of ≤0.8 is indicated on both graphs. Please click here to view a larger version of this figure.

Figure 5: Validation of Chemical Genetic Interactions. Serial dilutions of several isolated mutant strains grown on DMSO Control (A), 10 µM 5-FU (B), or and 0.015% methyl methanesulfonate (MMS; C).
| GO Category | P-value | Genes identified (hits) | # hits | Total # of Genes in GO Category |
| tRNA wobble uridine modification [GO:0002098] | 2.24 × 10-6 | SIT4 NCS6 URM1 KTI12 IKI3 TUM1 ELP3 | 7 | 24 |
| transcription, DNA-dependent [GO:0006351] | 1.69 × 10-5 | PDR3 LDB7 HIR1 HAP3 ROX3 SPT7 SNF5 RPA14 SAC3 HMO1 NGG1 SPT3 IES6 AFT1 RAI1 STB5 SDS3 MET18 CTK2 RPB4 SWI3 RPA12 KTI12 ASH1 SWI6 IKI3 GCR2 POP2 LEO1 SSN3 SGF11 ELP3 | 32 | 540 |
| translation [GO:0006412] | 4.28 × 10-5 | RPL19B RPS11B FES1 RPS9B RPL31A RPL24A RPL7A RPS25A RPL26B RPL11B RPL27A RPL34B RPL14A TEF4 RPS29A RPL6A AEP1 RPS16A MRP7 RPS19B RPS19A RPS7A | 22 | 318 |
| tRNA wobble position uridine thiolation [GO:0002143] | 1.88 × 10-4 | NCS6 URM1 TUM1 | 3 | 5 |
| regulation of transcription, DNA-dependent [GO:0006355] | 4.98 × 10-4 | PDR3 LDB7 HIR1 HAP3 ROX3 SPT7 SNF5 SAC3 HMO1 NGG1 SPT3 IES6 AFT1 RAI1 STB5 SDS3 SWI3 KTI12 ASH1 SWI6 IKI3 GCR2 POP2 LEO1 SSN3 SGF11 ELP3 | 27 | 507 |
| protein urmylation [GO:0032447] | 6.33 × 10-4 | NCS6 URM1 URE2 | 3 | 7 |
| mRNA export from nucleus in response to heat stress [GO:0031990] | 2.04 × 10-3 | RPB4 NUP120 NUP133 | 3 | 10 |
| nuclear polyadenylation-dependent CUT catabolic process [GO:0071039] | 2.04 × 10-3 | AIR1 TRF5 MPP6 | 3 | 10 |
| tryptophan metabolic process [GO:0006568] | 2.15 × 10-3 | TRP5 TRP3 | 2 | 3 |
| early endosome to Golgi transport [GO:0034498] | 2.75 × 10-3 | ENT5 TCA17 RCY1 | 3 | 11 |
| ribosomal small subunit biogenesis [GO:0042274] | 3.63 × 10-3 | SAC3 LTV1 RPS19B RPS19A | 4 | 24 |
| chromatin modification [GO:0016568] | 3.64 × 10-3 | LDB7 HIR1 SPT7 NGG1 SPT3 SDS3 ASH1 SGF11 ELP3 | 9 | 114 |
| ATP metabolic process [GO:0046034] | 4.23 × 10-3 | VMA2 VMA1 | 2 | 4 |
| vacuolar proton-transporting V-type ATPase complex assembly [GO:0070072] | 4.23 × 10-3 | VMA21 VPH2 | 2 | 4 |
| chromosome localization [GO:0050000] | 4.23 × 10-3 | NUP120 NUP133 | 2 | 4 |
| mRNA transport [GO:0051028] | 4.59 × 10-3 | DHH1 SAC3 LOC1 KAP114 NUP120 NUP133 | 6 | 58 |
| vacuolar acidification [GO:0007035] | 4.89 × 10-3 | VMA2 VMA1 VMA5 VPH2 | 4 | 26 |
| rRNA export from nucleus [GO:0006407] | 5.63 × 10-3 | NUP120 NUP133 RPS19B RPS19A | 4 | 27 |
| endocytosis [GO:0006897] | 6.57 × 10-3 | SLA1 EDE1 CDC50 ART5 RCY1 VPS1 END3 | 7 | 82 |
| nuclear mRNA surveillance of mRNA 3'-end processing [GO:0071031] | 6.92 × 10-3 | AIR1 MPP6 | 2 | 5 |
| ncRNA polyadenylation [GO:0043629] | 6.92 × 10-3 | AIR1 TRF5 | 2 | 5 |
| nuclear polyadenylation-dependent snoRNA catabolic process [GO:0071036] | 6.92 × 10-3 | AIR1 TRF5 | 2 | 5 |
| nuclear polyadenylation-dependent snRNA catabolic process [GO:0071037] | 6.92 × 10-3 | AIR1 TRF5 | 2 | 5 |
| tryptophan biosynthetic process [GO:0000162] | 6.92 × 10-3 | TRP5 TRP3 | 2 | 5 |
| protein insertion into ER membrane [GO:0045048] | 6.92 × 10-3 | GET1 GET4 | 2 | 5 |
| mRNA stabilization [GO:0048255] | 6.92 × 10-3 | ATP25 IGO1 | 2 | 5 |
| response to DNA damage stimulus [GO:0006974] | 7.05 × 10-3 | MUS81 RAD2 MET18 CTK2 RPB4 GRR1 DEF1 MMS22 RAD52 MRE11 RAD50 RMI1 | 12 | 197 |
| GO Category | P-value | Genes identified (hits) | # hits | Total # of Genes in GO Category |
| nuclear polyadenylation-dependent rRNA catabolic process [GO:0071035] | 8.46 × 10-3 | AIR1 TRF5 MPP6 | 3 | 16 |
Table 1: Gene ontology (GO) Characterization of 5-Fluorouracil Sensitive Mutants.
