

Summary
Protein aggregation elicits cellular oxidative stress. This protocol describes a method for monitoring the intracellular states of amyloidogenic proteins and the oxidative stress associated with them, using flow cytometry. The approach is used to study the behavior of soluble and aggregation-prone variants of the amyloid-β peptide.
Abstract
Protein misfolding and aggregation into amyloid conformations have been related to the onset and progression of several neurodegenerative diseases. However, there is still little information about how insoluble protein aggregates exert their toxic effects in vivo. Simple prokaryotic and eukaryotic model organisms, such as bacteria and yeast, have contributed significantly to our present understanding of the mechanisms behind the intracellular amyloid formation, aggregates propagation, and toxicity. In this protocol, the use of yeast is described as a model to dissect the relationship between the formation of protein aggregates and their impact on cellular oxidative stress. The method combines the detection of the intracellular soluble/aggregated state of an amyloidogenic protein with the quantification of the cellular oxidative damage resulting from its expression using flow cytometry (FC). This approach is simple, fast, and quantitative. The study illustrates the technique by correlating the cellular oxidative stress caused by a large set of amyloid-β peptide variants with their respective intrinsic aggregation propensities.
Introduction
Proteostasis is a fundamental determinant of cell fitness and aging processes. In cells, protein homeostasis is maintained by sophisticated protein quality control networks aimed to ensure the correct refolding of misfolded protein conformers by chaperones and/or their targeted proteolysis with several well-conserved mechanisms1,2,3,4,5. A large number of studies provide support to the link between the onset and progression of a broad range of human diseases and the failure of proteostasis, leading to protein misfolding and aggregation. For instance, the presence of protein deposits is considered a pathological hallmark of many neurodegenerative disorders, such as Alzheimer's, Parkinson's, and Huntington's diseases6,7,8, prionogenic diseases, and non-degenerative amyloidoses9. It has been suggested that early oligomeric and protofibrillar assemblies in the aggregation reaction are the main elicitors of cytotoxicity, establishing aberrant interactions with other proteins in the crowded cellular milieu10. In addition, protein inclusions (PI) can be transmitted between cells, propagating their toxic effect11,12. Therefore, it could be that the formation of PI might indeed constitute a detoxifying mechanism that restricts the presence of dangerous aggregated species to specific locations in the cell, where they can be processed or accumulated without major side effects13,14.
Standard in vitro biochemical approaches have provided important insights into the different species that populate aggregation reactions and their properties15,16. However, the conditions used in these assays are clearly different from those occurring within the cell and, therefore, question their physiological relevance. Because of the notable conservation of cellular pathways such as protein quality control, autophagy, or the regulation of the cellular redox state17,18 among eukaryotes19,20,21,22,23, the budding yeast Saccharomyces cerevisiae (S. cerevisiae) has emerged as a privileged simple cellular model to study the molecular determinants of protein aggregation and its associated cytotoxic impact in biologically relevant environments24,25,26.
Protein aggregation propensity is a feature inherently encoded in the primary sequence. Thus, the formation of amyloid-like structures can be predicted based on the identification and evaluation of the potency of aggregation-promoting regions in polypeptides27. However, despite the success of bioinformatic algorithms to predict the in vitro aggregation properties of protein sequences, they are still far from forecasting how these propensities translate into in vivo cytotoxic impact. Studies that address the link between the aggregated state of a given protein and its associated cellular damage in a systematic manner may help to circumvent this computational limitation. This connection is addressed in the present study, taking advantage of a large set of variants of the amyloid-β peptide Aβ42 differing only in a single residue, but displaying a continuous range of aggregation propensities in vivo28. In particular, an FC-based approach to identify the conformational species accounting for the oxidative damage elicited by aggregation-prone proteins in yeast cells is described. The methodology provides many advantages such as simplicity, high-throughput capability, and accurate quantitative measurement. This approach made it possible to confirm that PI play a protective role against oxidative stress.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. S. cerevisiae Cultures and Protein Expression
Note: Aβ variants exhibit different relative aggregation propensities due to a mutation in a single residue at position 19 (Phe19) of the Aβ42 peptide (Figure 1A). These peptide variants are tagged with green fluorescent protein (GFP), which acts as an aggregation reporter (Figure 1)29.
- Transform plasmids encoding for 20 Aβ42-GFP variants into yeast cells with a BY4741 parental background (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0)30. Each plasmid encodes for an Aβ42 mutant fused to GFP by a GSSGSSG linker. The plasmids are pESC(-URA) and include the selectable marker URA329.
- Prepare the synthetic complete medium without uracil (SC-URA) with the following components: 1.9 g/L yeast nitrogen base without uracil, 5 g/L ammonium sulfate, and 900 mL of ddH2O combined with 100 mL of 20% (w/v) glucose.
- Put one colony of the transformed yeast cells into 20 mL of the SC-URA medium containing 2% glucose and grow the cultures overnight at 30 °C under agitation at 210 rpm.
- The next day, grow new yeast cells cultures by inoculating 100 µL of the overnight culture into 5 mL of fresh SC-URA medium.
- At an OD590 of 0.5, centrifuge the cultures at 3,000 x g for 4 min, discard the supernatant, and resuspend the cells in the same volume of fresh SC-URA medium containing 2% raffinose.
- Incubate at 30 °C under agitation at 210 rpm for 30 min. Then, centrifuge the cells at 3,000 x g for 4 min and discard the supernatant. Resuspend the cells in fresh SC-URA medium containing 2% galactose to induce the recombinant protein expression.
- After 16 h of inducing protein expression in SC-URA medium containing 2% galactose, harvest 1 mL of the induced cells in sterile microcentrifuge tubes by centrifugation at 3,000 x g for 4 min.
NOTE: The most relevant differences occur after an induction period of 16 h. Induced yeast cells expressing Aβ42-GFP variants can be visualized by fluorescence microscopy to determine the recombinant protein distribution inside the cells (Figure 2B).
2. Cell Staining
Note: Fresh non-induced cells are required as a negative control to establish a fluorescent threshold during an FC analysis.
- Take the 16 h-induced yeast cells and determine their optical density at 590 nm (OD590). Dilute them in a sterile phosphate buffered saline (PBS) to an OD590 of 0.1.
- Prepare 1x phosphate buffered saline as follows: add 8 g of NaCl, 0.2 g of KCl, 1.44 g of Na2HPO4, and 0.24 g of KH2PO4 to 800 mL of ddH2O. Fill the solution up to 1 L of ddH2O after adjusting the pH to 7.4 with HCl. Filter the buffer through a 0.2 µm filter.
- Transfer the expressing cell suspensions to appropriately labeled tubes (12 × 75 mm round-bottom polystyrene) and protect them from light. Make sure they are ready to be loaded on the flow cytometer for analysis, together with non-induced and non-stained cells.
- Add the oxidative stress probe (e.g., CellROX Deep Red) to each sample at a final concentration of 5 µM and incubate the cells at 30 °C for 30 min in the dark.
- After incubation, wash the cells 3 times with 1x PBS and resuspend them in the same volume of buffer before their analysis by FC.
3. Flow Cytometry Analysis
Note: Use an FC setup with appropriate lasers and filters to detect GFP and oxidative stress probe fluorescence signal. A flow cytometer equipped with a 488 nm blue laser for the detection of GFP and a 635 nm red laser for the detection of oxidative stress probe fluorescence can be used. Acquisition of emission fluorescence of GFP and oxidative stress probe is performed with a 530/30 nm BP filter and a 660/20 BP filter, respectively (Table 1).
- Click on Open New Worksheet Panel and create the following acquisition dot plots.
- Select the Scatter Plot Tool from the toolbar and create a plot with the variables Side Scatter-Area (SSC-A) on the y-axis vs. Forward Scatter-Area (FSC-A) in a linear scale on the x-axis.
- Click the Scatter Plot Tool from the toolbar and create a plot with the variables FL1-Area (FITC) on the x-axis vs. FL3-Area (APC) in a logarithmic scale on the y-axis.
- Click the Instruments Settings icon. Click the Compensation tab and set all compensation levels. Click on the Acquisition tab and select a total number of 20,000 events to be recorded.
- Click the Flow Rate icon to switch the flow rate to Low.
- Click the Acquire tab to start running the non-induced, unstained cells and adjust the voltage in the Instrument Settings of the forward and side scatter until the population is distributed in the middle-left quadrant.
- Click on the Polygon icon to set a region R1 around the cell population excluding cell debris, and use this gated population P1 = R1 for all fluorescent dot plots and histogram representations (Figure 3).
- To adjust the PMT voltage of the fluorescence signal, run the unstained cells in the FL1-FL3 dot plot, tuning the gain in the Instrument Setting tab, until the cells are distributed in the lower left quadrant.
- Change the sample to the induced cells to measure GFP fluorescence. In the Instrument Setting tab, set the gain in the FSC-A vs. FL1 (FITC) when the population is distributed in the lower-right quadrant. Define the positive cell population with a gate (Gate P2).
- Change the sample to the non-induced stained cells to display oxidative stress by the fluorescence on an FSC-A vs. FL3 (APC) dot plot. Tune the gain until the cell population is distributed in the left-upper quadrant. Gate the positive cell population in P3.
- Make two histogram plots with the histogram icon to represent the cell fluorescence. To represent the fluorescence intensity, make sure FL1 (FITC) representing the GFP fluorescence is in the x-axis, and FL3 (APC) representing the fluorescence is in the y-axis, applying P2 and P3 populations on a logarithmic scale, respectively.
NOTE: Histogram overlays are a good way to illustrate differences between the fluorescence profiles of cells expressing different Aβ variants.
4. Recombinant Protein Immunodetection
- To determine protein expression levels, harvest the yeast cultures induced for 16 h by centrifugation at 4,000 x g for 6 min, and store the cell pellets at -80 °C.
- Resuspend the collected cells expressing recombinant protein for 16 h in PBS. Prepare 200 µL cell suspensions of each Aβ42-GFP mutant to an OD590 of 20.
- For total protein fraction analysis, harvest 100 µL of each mutant by centrifugation at 14,000 x g for 20 min and resuspend the cell pellets in the same volume of yeast lysis buffer Y-PER (50 mM Tris-HCl pH 8.0, 1% DMSO, 200 mM NaCl, 1 mM EDTA containing 10 mg/mL SB3-14 (myristyl sulfobetaine) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF)).
- Incubate the samples for 20 min at room temperature under mild agitation.
- Determine the protein extract concentration by using the Bradford assay. Load up to 5 µg of protein extract of each sample on a 15% acrylamide SDS-PAGE electrophoresis gel and blot onto a polyvinylidene difluoride (PVDF) membrane at 100 V for 60 min.
- Prepare a Bradford reagent with 100 mg of Coomassie Brilliant Blue G-250 dissolved in 50 mL of 95% ethanol and add 100 mL of 85% (w/v) phosphoric acid. Then, when the dye is completely dissolved, dilute the mixture with ddH2O to 1 L and filter it through cellulose filter paper just before use.
NOTE: The Bradford reagent should be light brown. - Prepare a standard of bovine serum albumin (BSA) ranging from 5 - 100 µg of protein in 1,000 µL of the Bradford reagent.
- Add 1 - 10 µL of protein extract to 1 mL of the Bradford reagent and incubate the mixture at room temperature for 5 min.
- Measure the absorbance of the standards and the protein extract samples at OD595.
- For the analysis, make a plot with the absorbance values obtained for the standards vs. µg of protein. Based on the standard curve, determine the concentrations of the original samples from the amount of protein, considering the volume and dilution, if any.
- Prepare a Bradford reagent with 100 mg of Coomassie Brilliant Blue G-250 dissolved in 50 mL of 95% ethanol and add 100 mL of 85% (w/v) phosphoric acid. Then, when the dye is completely dissolved, dilute the mixture with ddH2O to 1 L and filter it through cellulose filter paper just before use.
- Rinse the membrane with 1x Tris-buffered saline (TBS), 0.1% Tween 20 (TTBS), and block it using 5% (w/v) nonfat dry milk in 1x TTBS.
- To prepare 1 L of 10x TBS, dissolve 24 g of Tris base and 88 g of NaCl in 900 mL of H2Odd and adjust the pH to 7.6. Add ddH2O to a final volume of 1 L. For a 1x solution, mix 1 part of 10x stock with 9 parts of ddH2O.
- Incubate the membrane for 1 h with a primary β-amyloid antibody 6E10 diluted 1:1,000 and for 1 h at room temperature with a secondary antibody goat anti-mouse IgG-HRP conjugate diluted 1:10,000.
- Develop membranes using 1 mL of Luminata fluorescent reagent and quantify bands by densitometric analysis as described in step 5.2.
5. Data Analysis
- To analyze the data obtained by FC, create a table displaying the mean fluorescent intensity (MFI) and median fluorescence with its corresponding standard error and/or the coefficient of variance (CV) for GFP fluorescence and oxidative stress levels.
- Click the Inspector tab and the Statistics icon to customize the statistical data that are required for each channel.
NOTE: When comparing protein variants, convert fluorescence values data to a fold-over background or resolution metric (RD):
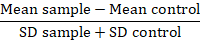
In this metric, the mean sample is the mean fluorescence of problem samples, the mean control is the mean fluorescence of cells used as a control, and the SD is the standard deviation of the mean fluorescence. RD factor is a more accurate index than MFI value as it not only calculates the difference between samples but also accounts for the spreading of the data. This calculation is specifically relevant when comparing data coming from different experiments carried out over time. Once the RDs have been calculated, the use of a statistical test is advisable to confirm the significance of the observed differences between protein variants.
- Click the Inspector tab and the Statistics icon to customize the statistical data that are required for each channel.
- For protein immunodetection, quantify the recombinant protein levels by Western blot using ImageJ software.
- Before the densitometry analysis, convert the original raw images of the developed membrane into JPEG file format and greyscale mode.
- Adjust the Set Measurements section of the Analyze menu to Mean Grey Value.
- Click on the Rectangle tool from ImageJ and define a region of interest (ROI) with a consistent size for the band densitometry analysis. Center each band on the membrane image inside the created rectangular frame and record the measurement one by one using Ctrl+M (the measurement command).
- Measure the local background adjacent to each band, applying the same ROI area. After measurement, subtract the corresponding local background from each individual band.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
This protocol describes how to employ a collection of 20 variants of the Aβ42 peptide where Phe19 has been mutated to all natural proteinogenic amino acids28. The theoretical aggregation propensities of these proteins can be analyzed using two different bioinformatic algorithms (AGGRESCAN and TANGO31,32). In both cases, this analysis renders a progressive gradation of aggregation tendencies, ascribing, as a general rule, the highest values to hydrophobic residues and the lowest to charged and polar ones (Figure 1B, 1C).
To track experimentally the intracellular aggregation state of different Aβ42 variants, the described experiment requires a transformation of a plasmid, encoding for Aβ42 and fused to GFP (Figure 1A) under the control of the galactose-inducible GAL1, into S. cerevisiae. Yeast cells expressing Aβ42 variants after an induction period of 16 h can be visualized under a fluorescence microscope. This visualization makes it possible to confirm the formation of PI in 10 out of the 20 variants from the analyzed collection. The number and size of the fluorescent aggregates differ from variant to variant (Figure 2). Figure 2A shows the PI quantification from a total of 500 cells in each of the two independent cultures. Here, we can observe an excellent agreement between predicted and in vivo aggregation properties.
To address whether the formation of protein aggregates can trigger oxidative stress in this model cell system, this protocol uses FC to monitor the cellular GFP fluorescence together with the fluorescence of the fluorogenic probe as an indicator of reactive oxygen species (ROS) production. Figure 3 illustrates the procedure and obtained results for Gln and Ile mutants, which correspond to the homogeneously distributed and PI-forming Aβ42 variants, respectively.
First, the analyzed cell population is gated (P1) in the FSC-A vs. SSC-A dot plot to remove cell detritus from the analysis (Figure 3A). The fluorescence signal of GFP vs. oxidative stress probe of the P1 population is represented in dot plot graphs where green fluorescent cells are gated in P3 (Figure 3B). Figure 3C and 3D show the histograms created to obtain statistical data for each expressed variant, including the CV (Table 2), in order to quantify the mean and the median fluorescence of each fluorescent marker.
All mutants are expected to be expressed at the same levels since they differ in a single amino acid (0.02% of the Aβ42-GFP sequence). However, the proteostasis machinery can respond in a differential manner to their particular aggregation propensities. Therefore, the protein expression levels in cellular extracts are quantified using an Aβ-specific antibody (Figure 4). We see that, as a general trend, PI-forming Aβ42 variants are present at lower levels than those remaining homogeneously distributed in the cytosol.
Important differences among Aβ42 variants can be observed when their oxidative stress probe fluorescence, protein levels, and GFP fluorescence properties are represented relative to their ability to form PI and their intrinsic aggregation propensities (Figure 5). The more aggregation-prone PI-forming variants elicit much lower oxidative stress than their more soluble counterparts (Figure 5A, 5B). With these results, there is no obvious correlation between the size and the number of aggregates per individual cell (Figure 2) and oxidative stress levels. PI-forming variants, especially mutants bearing Trp and Phe at position 19, are present in the cell at lower levels than those homogenously distributed in the cytosol (Figure 5C, 5D).The exception to this is the Thr mutant, for which less than 10% of the cells form PI (Figure 1B, 1C). This corresponds with the fact that the most aggregation-prone variants are selectively cleared by the yeast quality control degradation machinery28. GFP fluorescence and protein levels correlate well for the more soluble Aβ42 forms, but not for those forming PI (Figure 5E, 5F). This is probably because in these cellular populations, the fluorescence comes from two different compartments, the cytosol and the inclusions, and their relative contributions to the total fluorescence differ between mutants34.

Figure 1. Aggregation propensity analysis for the collection of Aβ42-GFP fusion constructs derived from the mutation of residue at position 19 in the Aβ42 peptide by all natural amino acids. A. This image shows a 3D model of wt Aβ42 fused to GFP by a linker (Aβ42, grey; GFP, green), based on the PDB 1EMA for green fluorescent protein from Aequorea victoria and the 2OTK for the Alzheimer Aβ peptide in which the Phe19 side chain of wt Aβ42 is shown in red. The model is created using Pymol software. Residue 19 in the Aβ42 sequence occupies a central position in the central hydrophobic cluster. The bar graphs are created with two different bioinformatic predictors: B. AGGRESCAN, C. TANGO. Please click here to view a larger version of this figure.

Figure 2. Aβ42-GFP PI-forming variants in S. cerevisiae cultures induced for 16 h of expression. A. The bar graph indicates the percentage of cells containing a different number of PI calculated from a total of 500 fluorescent cells for each variant in two biological replicates. B. These images are representative fluorescent microscopy images of selected Aβ42-GFP variants (Phe, Ile, Thr, Trp). They were acquired under UV light using an excitation filter for GFP (450 - 500 nm) and an emission range (515 - 560 nm). The scale bar represents 10 µm. Please click here to view a larger version of this figure.

Figure 3. Scheme for flow cytometry (FC) analysis of S. cerevisiae cells expressing selected Aβ42-GFP variants. A. The chart shows gated yeast cells (P1) in dot plots (FSC-A vs. SSC-A) where cell detritus is removed from the cell population. The microscopic images of Gln (homogeneously distributed) and Ile (PI-forming) variants are shown next to the FC dot plots. The scale bar represents 10 µm. B. These scatter dot plot images represent GFP-A vs. oxidative stress probe in which the gated population (P3) includes only fluorescent cells, excluding the background signal. The cell frequency histograms are of C. the GFP signal (FITC amplitude) gated from P1 and D. CellROX (APC amplitude) gated from P3. The cell acquisition was performed with a flow cytometer. Each plot represents 20,000 events. Q and I correspond to Gln and Ile Aβ42 mutants, respectively. Please click here to view a larger version of this figure.

Figure 4. Quantification of cellular protein levels. This figure shows Western blots of the total protein fractions of Aβ42-GFP mutants after 16 h of expression in S. cerevisiae. The PI-forming variants are colored in green and those diffusely distributed in the cytosol are colored in light red. Please click here to view a larger version of this figure.

Figure 5. Cellular oxidative stress, intracellular GFP fluorescence, and cellular protein levels for Aβ42-GFP mutants determined after 16 h of expression in S. cerevisiae. The variants represented on the x-axis have been ordered according to their predicted aggregation propensities by either AGGRESCAN (left panel) or TANGO (right panel). The PI-forming variants are colored in green and the non-PI-forming variants are colored in light red. A. B. These bar graphs show the oxidative stress probe fluorescence values obtained by an FC analysis of the Aβ42-GFP mutants. C. D. These bar graphs represent the protein levels as quantified by a Western blot densitometry analysis using ImageJ software. E. F. These bar graphs show the GFP fluorescence values after an FC analysis. The error bars for the oxidative stress probe and GFP fluorescence values represent the coefficient of variance (CV) of the FC-gated cells. The error bars for the protein expression levels represent ± SE (n = 3). Please click here to view a larger version of this figure.
| Channel | Fluorophore | Excitation Wavelength (nm) | Band Pass Filter |
| FITC | GFP | 488 | 530/30 |
| APC | CellRox | 635 | 660/20 |
| PerCP | IP | 488 | 585/42 |
Table 1. Laser source, band-pass filters, and fluorophores used in flow cytometer.
| GFP fluorescence | CV | CellROX fluorescence | CV | |
| A | 9316 | 96.4 | 1964 | 127.5 |
| C | 9709 | 91.9 | 1275 | 172.2 |
| D | 11213 | 101.7 | 3443 | 155.8 |
| E | 12256 | 101.1 | 3220 | 152.2 |
| F | 3010 | 96.1 | 1245 | 146.4 |
| G | 11541 | 97.2 | 2947 | 158 |
| H | 7895 | 98.2 | 3582 | 120.8 |
| I | 7365 | 97.2 | 1416 | 141.3 |
| K | 10839 | 100.4 | 3102 | 122.1 |
| L | 7605 | 96.9 | 1401 | 161.8 |
| M | 8149 | 96 | 1308 | 170.4 |
| N | 12741 | 97.5 | 3403 | 134.9 |
| P | 9768 | 102.8 | 2629 | 143.6 |
| Q | 13066 | 91.3 | 3354 | 169.9 |
| R | 8537 | 101.3 | 2839 | 127.5 |
| S | 12053 | 99.1 | 3313 | 174.3 |
| T | 10615 | 97.7 | 2213 | 107.7 |
| V | 9169 | 96.1 | 1878 | 121.7 |
| W | 1715 | 94.7 | 1531 | 100 |
| Y | 7574 | 94.5 | 1234 | 138 |
Table 2. List of values for GFP fluorescence and oxidative stress probe fluorescence obtained by FC analysis of yeast cells expressing Aβ42-GFP mutants for 16 h. This table shows the mean fluorescence intensity (MFI) and the CV for each mutant.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
A wide range of diseases is linked to the accumulation of misfolded proteins into cellular deposits6,7,8,33. Many efforts have been made to unravel the molecular mechanisms that trigger the onset of these diseases using computational approaches, which do not take into account protein concentrations, or in vitro approaches, in which the protein concentration remains constant during the reaction. However, within the cell, proteins are constantly synthesized and degraded in a crowded and non-homogeneous environment. This explains the frequent discrepancies between in silico, in vitro, and in vivo aggregation properties of amyloidogenic proteins.
There are multiple reasons why simple cellular models, such as yeast, are a reasonable choice to study the aggregation and associated toxicity of proteins related to neurodegenerative diseases in a more biological context. For instance, they give us the possibility to analyze the impact of protein misfolding aggregation on an intact cellular population using fast analyses such as the one implemented here. However, we should consider that oxidative levels may differ among yeast strains. Thus, to compare between strains, or even between different proteins, oxidants and reductants, such as DTT and diamine, should be used to establish an oxidation/reduction scale.
In the example illustrated in this article, the bioinformatic analysis, cellular protein quantification, protein localization imaging and simultaneous FC analysis of GFP activity and oxidative stress levels have been integrated to identify the molecular species responsible for the oxidative damage elicited by protein aggregation reactions. The results demonstrate that the more soluble Aβ42 variants, rather than the more aggregation-prone, promote the highest oxidative stress. This points to the diffusible species being the more dangerous Aβ42 species in vivo. The lower toxicity of aggregating variants seems to respond both to the fact that these conformations are sequestered into PI and to their preferential degradation by the protein quality machinery, resulting in lower protein levels than that of their soluble counterparts.
The described method is not limited to the analysis of the oxidative stress produced by Aβ42 aggregated/soluble species and can also be applied to study a variety of protein aggregation disorders. Furthermore, the technique might be useful to monitor the effect of aggregation inhibitors on cellular oxidative stress, allowing to discard for further clinical applications those molecules that promote the accumulation of oxidative active protein species. Finally, as long as a fluorogenic probe is available, the approach offers remarkable opportunities to identify the molecular species responsible for other toxic effects linked to protein aggregation in a fast an easy way, providing quantitative data.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Materials
| Name | Company | Catalog Number | Comments |
| Yeast cells BY4741 | ATCC | 201388 | Genotype: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| pESC(-Ura) plasmid | Agilent Genomics | 217454 | Yeast expression plasmid with a Gal promotor. Selectable marker URA3 |
| Yeast Synthetic Drop-out Medium Supplements | Sigma | Y1501 | Powder |
| Yeast Nitrogen Base Without Amino Acids | Sigma | Y0626 | Powder |
| Raffinose | Sigma | R7630 | Powder |
| Glucose | Sigma | G7021 | Powder |
| Galactose | Sigma | G0750 | Powder |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | Solution 10X |
| CellROX Deep Red Reagent | Life Technologies | C10422 | Free radical cell-permeant fluorescent sensor, non-fluorescent while in a reduced state, and exhibits bright fluorescence upon oxidation by reactive oxygen species (ROS), with absorption/emission maxima at 644/665 nm. |
| Y-PER protein extraction reagent | Thermo Scientific | 78990 | Liquid cell lysis buffer |
| Acrylamide/Bis-acrylamide | Sigma | A6050 | Solution |
| Bradford dye reagent | Bio-Rad | 5000205 | Dye reagent for one-step determination of protein concentration |
| β-amyloid antibody 6E10 | BioLegend | 803001 | Mouse IgG1. The epitope lies within amino acids 3-8 of beta amyloid (EFRHDS). |
| Goat anti-mouse IgG-HRP conjugate | Bio-Rad | 1721011 | |
| Membrane Immobilon-P, PVDF | Millipore | IPVH00010 | |
| Luminata forte | Merk | WBLUF0100 | Premixed, ready to use chemiluminescent HRP detection reagent |
| Phenylmethanesulfonyl fluoride solution (PMSF) | Sigma | 93482 | Protease inhibitor. Dissolved at 0.1 M in ethanol |
| FACSCanto flow cytometer | BD Biosciences | 657338 | Equipped with a 488 nm blue laser for the detection of GFP, and 635 nm red laser / 530/30 nm BP filter and 660/20 BP filter |
| Mini Trans-Blot Electrophoresis Transfer cell | Bio-Rad | 1703930 | Protein transference system |
| Mini-PROTEAN Tetra Handcast Systems | Bio-Rad | 1658000FC | Electrophoresis system |
References
- Frydman, J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annual Review of Biochemistry. 70, 603-647 (2001).
- Hartl, F. U., Bracher, A., Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature. 475, 324-332 (2011).
- Wong, E., et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nature Communications. 3, (2012).
- Winkler, J., Tyedmers, J., Bukau, B., Mogk, A. Chaperone networks in protein disaggregation and prion propagation. Journal of Structural Biology. 179, 152-160 (2012).
- Glickman, M. H., Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological Reviews. 82, 373-428 (2002).
- Dobson, C. M. Getting out of shape. Nature. 418, 729-730 (2002).
- Chiti, F., Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annual Review of Biochemistry. 75, 333-366 (2006).
- Aguzzi, A., O'Connor, T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery. 9, 237-248 (2010).
- Rapezzi, C., et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nature Reviews Cardiology. 7, 398-408 (2010).
- Deas, E., et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxidants & Redox Signaling. 24, 376-391 (2016).
- Brundin, P., Melki, R., Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature Reviews Molecular Cell Biology. 11, 301-307 (2010).
- Frost, B., Diamond, M. I. Prion-like mechanisms in neurodegenerative diseases. Nature Reviews Neuroscience. 11, 155-159 (2009).
- Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431, 805-810 (2004).
- Ross, C. A., Poirier, M. A. Opinion: what is the role of protein aggregation in neurodegeneration? Nature Reviews Molecular Cell Biology. 6, 891-898 (2005).
- Mishra, R., Sjölander, D., Hammarström, P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Molecular BioSystems. 7, 1232-1240 (2011).
- Klunk, W. E., Jacob, R. F., Mason, R. P. Quantifying amyloid by congo red spectral shift assay. Methods in Enzymology. 309, 285-305 (1999).
- Khurana, V., Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nature Reviews Neuroscience. 11, 436-449 (2010).
- Tenreiro, S., Outeiro, T. F. Simple is good: yeast models of neurodegeneration. FEMS Yeast Research. 10, 970-979 (2010).
- Moosavi, B., Mousavi, B., Macreadie, I. G. Yeast model of amyloid-β and Tau aggregation in Alzheimer's disease. Journal of Alzheimer's Disease. 47, 9-16 (2015).
- Tenreiro, S., Munder, M. C., Alberti, S., Outeiro, T. F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. Journal of Neurochemistry. 127, 438-452 (2013).
- Yang, J., Hao, X., Cao, X., Liu, B., Nyström, T. Spatial sequestration and detoxification of huntingtin by the ribosome quality control complex. eLife. 5, (2016).
- Braun, R. J., Büttner, S., Ring, J., Kroemer, G., Madeo, F. Nervous yeast: modeling neurotoxic cell death. Trends in Biochemical Sciences. 35, 135-144 (2010).
- Figley, M. D., Gitler, A. D. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Diseases. 1, e24420 (2013).
- Cooper, A. A., et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 313, 324-328 (2006).
- Johnson, B. S., McCaffery, J. M., Lindquist, S., Gitler, A. D. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 105, 6439-6444 (2008).
- Bharathi, V., et al. Use of ade1 and ade2 mutations for development of a versatile red/white color assay of amyloid-induced oxidative stress in saccharomyces cerevisiae. Yeast. 33, 607-620 (2016).
- Pallarès, I., Ventura, S. Advances in the prediction of protein aggregation propensity. Current Medicinal Chemistry. , (2017).
- Villar-Piqué, A., Ventura, S. Protein aggregation propensity is a crucial determinant of intracellular inclusion formation and quality control degradation. Biochimica et Biophysica Acta. 1833, 2714-2724 (2013).
- Navarro, S., Villar-Piqué, A., Ventura, S. Selection against toxic aggregation-prone protein sequences in bacteria. Biochimica et Biophysica Acta. 1843, 866-874 (2014).
- Morell, M., de Groot, N. S., Vendrell, J., Avilés, F. X., Ventura, S. Linking amyloid protein aggregation and yeast survival. Molecular BioSystems. 7, 1121-1128 (2011).
- Conchillo-Solé, O., et al. AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics. 8, 65 (2007).
- Fernandez-Escamilla, A. M., Rousseau, F., Schymkowitz, J., Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology. 22, 1302-1306 (2004).
- Renner, M., Melki, R. Protein aggregation and prionopathies. Pathologie Biologie.(Paris). 62, 162-168 (2014).
- Carija, A., Navarro, S., de Groot, N. S., Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biology. 12, 699-711 (2017).
