

Summary
Proteinaggregation entlockt zellulären oxidativen Stress. Dieses Protokoll beschreibt eine Methode zur Überwachung der intrazellulären Staaten von Amyloidogenic Proteinen und den oxidativen Stress zugeordnet werden, mit Hilfe der Durchflusszytometrie. Der Ansatz dient zur Untersuchung des Verhaltens von löslichen und Aggregation neigende Varianten des β-Amyloid Peptids.
Abstract
Protein misfolding und Aggregation in Amyloid Konformationen haben an der Entstehung und Progression von mehreren neurodegenerativen Erkrankungen in Zusammenhang gebracht. Allerdings gibt es noch wenig Informationen über wie unlöslichen Protein Aggregate ihre toxische Wirkung in Vivoausüben. Einfaches Modell der prokaryotischen und eukaryotischen Organismen, wie Bakterien und Hefen, trugen wesentlich zur unsere gegenwärtigen Verständnis der Mechanismen hinter der intrazellulären Amyloid Bildung, Aggregate Ausbreitung und Toxizität. In diesem Protokoll bezeichnet man die Verwendung von Hefe als ein Modell, um die Beziehung zwischen der Entstehung von Protein-Aggregate und ihre Auswirkungen auf die zellulären oxidativen Stress zu sezieren. Die Methode verbindet die Erkennung der intrazellulären löslich/aggregiert Bundesstaat ein Amyloidogenic Protein mit der Quantifizierung der zellulären oxidativen Schäden, die aus ihren Ausdruck mit Durchflusszytometrie (FC). Dieser Ansatz ist einfach, schnell und quantitative. Die Studie zeigt die Technik durch Korrelation der zellulären oxidativen Stress, verursacht durch eine große Anzahl von Amyloid-β-Peptid-Varianten mit ihren jeweiligen inneren Aggregation Neigungen.
Introduction
Proteostase ist eine wesentliche Determinante des Zellprozesse Fitness und Alterung. In den Zellen bleibt Protein Homöostase durch anspruchsvolle Protein-Qualitätskontrolle, die Netzwerke zielte darauf ab, sicherzustellen, das richtige umfaltung fehlgefaltete Protein Konformere durch Chaperone und/oder ihre gezielten Proteolyse mit mehreren gut erhaltenen Mechanismen1 ,2,3,4,5. Eine große Anzahl von Studien unterstützen die Verbindung zwischen der Entstehung und Progression von ein breites Spektrum von Erkrankungen des Menschen und das Scheitern der proteostase, führt zu Protein misfolding und Aggregation. Zum Beispiel gilt das Vorhandensein von Proteinablagerungen eine pathologische Gütesiegel von vielen neurodegenerativen Erkrankungen wie Alzheimer, Parkinson und Huntington Krankheiten6,7,8, Prionogenic Krankheiten, und nicht-degenerativen Amyloidosen9. Es wurde vermutet, dass frühe Oligomere und Protofibrillar Baugruppen in der Aggregation Reaktion die wichtigsten spüren der Zytotoxizität sind, aberrante Interaktionen mit anderen Proteinen in den überfüllten zellulären Milieu10Aufbau. Darüber hinaus können Protein Einschlüsse (PI) zwischen den Zellen, verbreiten ihre toxische Wirkung11,12übertragen werden. Daher könnte es sein, dass die Bildung von PI in der Tat einen entgiftenden Mechanismus, der das Vorhandensein von gefährlichen aggregierten Arten zu bestimmten Stellen in der Zelle beschränkt darstellen, wo sie verarbeitet oder angesammelt ohne große Nebenwirkungen 13 , 14.
Standard in-vitro- biochemische Ansätze lieferten wichtige Einblicke in die verschiedenen Arten, die Aggregation Reaktionen und ihre Eigenschaften15,16bevölkern. Jedoch Bedingungen verwendet in diesen Tests unterscheiden sich deutlich von denen, die innerhalb der Zelle und ihre physiologische Bedeutung daher in Frage zu stellen. Aufgrund der bemerkenswerten Erhaltung zelluläre Signalwege wie Protein-Qualitätskontrolle, Autophagie oder die Regulierung der zellulären Redox Zustand17,18 unter Eukaryoten19,20,21 ,22,23, die angehende Hefe Saccharomyces Cerevisiae (S. Cerevisiae) entstanden als privilegierte einfaches zellulären Modell, die molekulare Determinanten der Proteinaggregation zu studieren und seiner zugehörigen zytotoxische Wirkung auf biologisch relevante Umgebungen24,25,26.
Protein Aggregation Neigung ist eine Funktion, die von Natur aus in der primären Sequenz codiert. So kann die Bildung der Amyloid-ähnliche Strukturen vorhergesagt werden, basierend auf die Identifikation und Bewertung der Potenz der Aggregation-Förderung Regionen in Polypeptide27. Trotz des Erfolgs der bioinformatische Algorithmen, die in-vitro- Aggregation-Eigenschaften von Proteinsequenzen vorherzusagen, sind sie jedoch noch lange nicht vorhersagen, wie diese Neigungen in in Vivo zytotoxische Wirkung zu übersetzen. Studien, die die Verbindung zwischen den aggregierten Zustand eines gegebenen Proteins und seiner damit verbundenen Zellschädigung in systematischer Weise ansprechen können helfen, um diese rechnerische Einschränkung zu umgehen. Diese Verbindung ist in der vorliegenden Studie, unter Ausnutzung von einer großen Anzahl von Varianten des β-Amyloid Peptids Aβ42 unterscheiden sich nur in einer einzigen Rückstand, aber Anzeige einen fortlaufenden Bereich von Aggregation Neigungen in Vivo28gerichtet. Insbesondere bezeichnet man ein FC-Ansatz für die Konformationsänderungen Arten entfallen die oxidativen Schäden hervorgerufen durch Aggregation neigen Proteine in Hefezellen zu identifizieren. Die Methode bietet viele Vorteile wie Einfachheit, Hochdurchsatz-Fähigkeit und genaue quantitative Messung. Dieser Ansatz ermöglichte es, dass PI eine schützende Rolle gegen oxidativen Stress spielen zu bestätigen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. S. Cerevisiae Kulturen und Protein-Expression
Hinweis: Aβ-Varianten weisen unterschiedliche relative Aggregation Neigungen aufgrund einer Mutation in einem einzigen Rückstand an Position 19 (Phe19) des Aβ42 Peptid (Abbildung 1A). Diese Peptid-Varianten sind mit grün fluoreszierenden Proteins (GFP), fungiert als eine Aggregation Reporter (Abbildung 1)29versehen.
- Plasmide, die Codierung für 20 Aβ42-GFP-Varianten in Hefezellen mit einem BY4741 elterlichen Hintergrund (MATa seine3Δ1 Leu2Δ0 traf15Δ0 ura3Δ0)30zu verwandeln. Jedes Plasmid kodiert für ein Aβ42 Mutant mit GFP durch einen GSSGSSG Linker verschmolzen. Plasmide sind pESC(-URA) und beinhalten die auswählbare Markierung URA329.
- Bereiten Sie das synthetische komplette Medium ohne Uracil (SC-URA) mit den folgenden Komponenten: 1,9 g/L Hefe Stickstoff base Uracil, 5 g/L Ammoniumsulfat und 900 mL DdH2O kombiniert mit 100 mL Glukose 20 % (w/V).
- Eine Kolonie der transformierten Hefezellen in 20 mL des Mediums SC-URA, enthält 2 % Glukose und wachsen Sie die Kulturen über Nacht bei 30 ° C unter Schütteln bei 210 u/min.
- Wachsen Sie am nächsten Tag neue Hefekulturen Zellen durch Impfkeimen 100 µL der Übernacht-Kultur in 5 mL frisches SC-URA-Medium.
- Bei einer OD590 von 0,5 die Kulturen bei 3.000 x g für 4 min zentrifugieren, überstand verwerfen und Aufschwemmen der Zellen in die gleiche Menge an frischer SC-URA-Speichermedium mit 2 % Raffinose.
- Bei 30 ° C unter Agitation bei 210 u/min für 30 min inkubieren. Dann die Zellen bei 3.000 x g für 4 min zentrifugieren und den überstand verwerfen. Aufzuwirbeln Sie die Zellen in frischen SC-URA-Speichermedium mit 2 % Galaktose induzieren die rekombinante Proteinexpression.
- Nach 16 h zur Induktion der Proteinexpression in SC-URA 2 % Galaktose-haltigem Medium Ernte 1 mL der induzierten Zellen in sterilen Mikrozentrifugenröhrchen durch Zentrifugation bei 3.000 x g für 4 Minuten.
Hinweis: Die wichtigsten Unterschiede auftreten, nachdem eine Einarbeitungszeit von 16 h. induzierte Hefezellen mit dem Ausdruck Aβ42-GFP-Varianten durch Fluoreszenz-Mikroskopie zur Bestimmung der rekombinanten PROTEINVERTEILUNG innerhalb der Zellen (Abb. 2 b) sichtbar gemacht werden kann.
(2) Zelle färben
Hinweis: Frischzellen-induzierte sind als Negativkontrolle herstellen eine fluoreszierende Schwelle während einer FC-Analyse erforderlich.
- Die 16 h-induzierte Hefezellen übernehmen und bestimmen ihre optische Dichte bei 590 nm (OD590). Verdünnen sie in eine sterile Phosphat gepufferte Kochsalzlösung (PBS) ein OD590 von 0,1.
- 1 x Phosphat gepufferte Kochsalzlösung wie folgt vorbereiten: 8 g NaCl, KCl, 1,44 g Na2HPO4, 0,2 g und 0,24 g KH2PO4 bis 800 mL DdH2O. füllen Sie die Lösung bis zu 1 L DdH2O nach Einstellen des pH-Werts auf 7,4 mit HCl. Filter des Puffers durch einen 0,2 µm-Filter.
- Transfer mit dem Ausdruck ihrer Zellsuspensionen entsprechend gekennzeichneten Rohre (12 × 75 mm Rundboden Polystyrol) und vor Licht zu schützen. Stellen Sie sicher, dass sie bereit sind, auf das Durchflusszytometer für Analyse, zusammen mit nicht-induzierte und nicht-gefärbten Zellen geladen werden.
- Jede Probe auf eine Endkonzentration von 5 µM die oxidativen Stress-Sonde (z. B. CellROX tiefrot) hinzu und inkubieren Sie die Zellen bei 30 ° C für 30 Minuten im Dunkeln.
- Waschen Sie nach der Inkubation der Zellen 3 Mal mit 1 X PBS und Aufschwemmen sie in das gleiche Volumen des Puffers vor ihrer Analyse von FC.
(3) Flow Cytometry Analysis
Hinweis: Verwenden Sie eine FC-Setup mit geeigneten Laser und Filter zur Erkennung von GFP und oxidativem Stress Sonde Fluoreszenzsignal. Ein Durchflusszytometer ausgestattet mit 488 nm blaue Laser für den Nachweis der GLP und einem 635 nm rot-Laser für den Nachweis von oxidativem Stress Sonde Fluoreszenz kann verwendet werden. Erwerb der Fluoreszenz Emission von GFP und oxidativem Stress Sonde erfolgt mit einem 530/30 nm BP und ein 660/20 BP Filter, bzw. (Tabelle 1).
- Klicken Sie auf Neues Arbeitsblatt Öffnungsklappe und erstellen Sie die folgenden Erwerb Punkt plottet.
- Wählen Sie aus der Symbolleiste das Scatter Plot-Tool und erstellen Sie ein Grundstück mit den Variablen Side Scatter-Area (SSC-A) auf der y-Achse vs. Forward Scatter-Bereich (FSC-A) in einer linearen Skala auf der x-Achse.
- Klicken Sie auf das Scatter Plot-Werkzeug aus der Werkzeugleiste und erstellen Sie ein Grundstück mit der Variablen FL1-Bereich (FITC) auf der x-Achse Vs. FL3-Bereich (APC) in einer logarithmischen Skala auf der y-Achse.
- Klicken Sie auf das Symbol " Instrumenten-Einstellungen ". Klicken Sie auf die Registerkarte " Entschädigung " und legen Sie alle Vergütungsstufen. Klicken Sie auf die Registerkarte " Erwerb " und wählen Sie eine Gesamtanzahl von 20.000 Ereignisse aufgezeichnet werden.
- Klicken Sie auf Durchfluss , um den Durchfluss zu niedrigzu wechseln.
- Klicken Sie auf die Registerkarte " Acquire " anfangen zu laufen-induzierte, ungefärbten Zellen, und passen Sie die Spannung in den Geräteeinstellungen des Forward und Side Scatter, bis die Bevölkerung in der Mitte-Links-Quadranten verteilt ist.
- Klicken Sie auf das Polygon -Symbol, um eine Region R1 um die Zellpopulation ausgenommen zellenrückstand gesetzt, und verwenden Sie diese geschlossene Population P1 = R1 für alle fluoreszierenden Punkt plottet und Histogramm Darstellungen (Abbildung 3).
- Um die PMT-Spannung des Fluoreszenzsignals anzupassen, führen Sie die ungefärbten Zellen im FL1-FL3 Dot Plot, den Gewinn in der Registerkarte " Geräteeinstellung " tuning, bis die Zellen im unteren linken Quadranten verteilt sind.
- Ändern der Probenmaterials auf die induzierte Zellen GFP Fluoreszenz zu messen. Stellen Sie im Register Geräteeinstellung Gain in der FSC-A vs. FL1 (FITC) Wenn die Bevölkerung im rechten unteren Quadranten verteilt wird. Definieren Sie die positiven Zellpopulation mit einem Tor (Tor P2).
- Veränderung der Probe, die nicht induzierten gefärbten Zellen oxidativen Stress durch die Fluoreszenz auf ein FSC-A vs. FL3 (APC) Dot-Plot angezeigt. Tunen Sie die Verstärkung, bis die Zellpopulation im linken oberen Quadranten verteilt wird. Tor der positiven Zellpopulation im P3.
- Machen Sie zwei Histogramm-Grundstücke mit dem Histogramm -Symbol um die Fluoreszenz der Zelle darzustellen. Der Fluoreszenzintensität darzustellen, stellen Sie sicher FL1 (FITC) repräsentieren die GFP Fluoreszenz ist auf der x-Achse und FL3 (APC) repräsentieren die Fluoreszenz in der y-Achse P2 und P3 Populationen bzw. auf einer logarithmischen Skala anwenden.
Hinweis: Histogramm Overlays sind eine gute Möglichkeit, Unterschiede zwischen den Fluoreszenz-Profilen von Zellen mit dem Ausdruck Aβ-Varianten zu verdeutlichen.
(4) rekombinante Protein Immunodetection
- Um proteingehalte Ausdruck zu ermitteln, Ernten Sie die Hefekulturen für 16 h durch Zentrifugation bei 4.000 x g für 6 min induziert, und speichern Sie die Zelle Pellets bei-80 ° C.
- Aufschwemmen der gesammelten Zellen rekombinantes Protein für 16 h mit PBS-Puffer zum Ausdruck zu bringen. 200 µL Zellsuspensionen jedes Aβ42-GFP mutierte zu einem OD-590 20 vorzubereiten.
- Für die Gesamt-Protein Bruchteil Analyse, Ernte 100 µL jeder Mutant durch Zentrifugation bei 14.000 x g für 20 min und Aufschwemmen der Zelle-Pellets in die gleiche Menge an Hefe Lyse Puffer Y-pro (50 mM Tris-HCl pH 8.0, 1 % DMSO, 200 mM NaCl, 1 mM EDTA enthalten 10 mg/mL SB3-14 (Myr Istyl Sulfobetaine) ergänzt mit 1 mM Phenylmethylsulfonyl Fluorid (PMSF)).
- Inkubieren Sie die Proben für 20 min bei Raumtemperatur unter milden Agitation.
- Bestimmen Sie die Proteinkonzentration Extrakt mithilfe des Bradford-Tests. Laden Sie bis zu 5 µg Proteinextrakt jeder Probe auf eine 15 % Acrylamid SDS-PAGE-Elektrophorese Gel und Blot auf eine Polyvinylidene Difluoride (PVDF) Membran bei 100 V 60 Minuten.
- Bereiten Sie ein Bradford Reagenz mit 100 mg Coomassie Brilliant Blue G-250 in 50 mL 95 % Ethanol gelöst und 100 mL Phosphorsäure 85 % (w/V). Dann, wenn der Farbstoff vollständig aufgelöst ist, verdünnen Sie die Mischung mit DdH2O, 1 L und Filtern sie durch Zellulose Filterpapier kurz vor Gebrauch.
Hinweis: Die Bradford-Reagenz sollte sein hellbraun. - Bereiten Sie einen Standard von Rinderserumalbumin (BSA) von 5-100 µg Protein in 1.000 µL des Bradford-Reagenz.
- Fügen Sie 1 bis 10 µL des Proteins zu 1 mL der Bradford-Reagenz zu extrahieren und die Mischung bei Raumtemperatur für 5 min inkubieren.
- Messung der Extinktion des Standards und die Proteinproben Extrakt bei OD595.
- Machen Sie für die Analyse ein Grundstück mit der Extinktion-Werte, die für die Normen Vs. µg Protein. Anhand der Standardkurve, bestimmen Sie die Konzentrationen an die original-Samples aus der Menge an Protein, wenn man bedenkt, das Volumen und die Verdünnung, falls vorhanden.
- Bereiten Sie ein Bradford Reagenz mit 100 mg Coomassie Brilliant Blue G-250 in 50 mL 95 % Ethanol gelöst und 100 mL Phosphorsäure 85 % (w/V). Dann, wenn der Farbstoff vollständig aufgelöst ist, verdünnen Sie die Mischung mit DdH2O, 1 L und Filtern sie durch Zellulose Filterpapier kurz vor Gebrauch.
- Spülen Sie die Membran mit 1 x Tris gepufferte Kochsalzlösung (TBS), 0,1 % Tween 20 (TTB), und es mit 5 % (w/V) fettfreie Trockenmilch in 1 X TTBS zu blockieren.
- Um 1 L 10 X TBS vorzubereiten, lösen Sie 24 g Tris-base und 88 g NaCl in 900 mL H2ODd auf und passen Sie den pH-Wert auf 7,6. Fügen Sie DdH2O zu einem Endvolumen von 1 L. Mischen Sie für eine Lösung 1 X 1 Teil 10 X Lager mit 9 Teile des DdH2O.
- Die Membran für 1 h mit einem β-Amyloid Primärantikörper 6E10 verdünnt 1:1,000 brüten und für 1 h bei Raumtemperatur mit einer Sekundärantikörper Ziege Anti-Maus IgG-HRP Konjugat verdünnt 01:10, 000.
- Entwickeln Sie Membranen mit 1 mL Luminata fluoreszierende Reagenz zu und quantifizieren Sie Bänder durch densitometrische Analysen zu, wie im Schritt 5.2 beschrieben.
(5) Datenanalyse
- Um die Daten, die von FC zu analysieren, erstellen Sie eine Tabelle zeigt die mittlere fluoreszente Intensität (MFI) und mittlere Fluoreszenz mit den entsprechenden Standard-Fehler und/oder der Koeffizient der Varianz (CV) für GFP Fluoreszenz und oxidativen Stress.
- Klicken Sie auf die Registerkarte " Inspektor " und die Statistik -Symbol, um die statistischen Daten anpassen, die für jeden Kanal erforderlich sind.
Hinweis: Beim Vergleich von Protein-Varianten konvertieren Sie Fluoreszenz Werte Daten zu einer Falte-Over Hintergrund oder Auflösung Metrik (RD):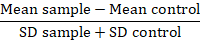
In dieser Metrik die mittlere Probe ist die mittlere Fluoreszenz Problem Proben, das mittlere Steuerelement ist die mittlere Fluoreszenz von Zellen als Steuerelement verwendet und die SD ist die Standardabweichung der mittleren Fluoreszenz. RD-Faktor ist ein genauer Index als MFI-Wert, da es nicht nur den Unterschied zwischen Proben, sondern auch Konten für die Verbreitung der Daten berechnet. Diese Berechnung ist besonders relevant, wenn Daten aus verschiedenen Experimenten vergleichen im Laufe der Zeit durchgeführt. Einmal das RDs berechnet wurden, empfiehlt die Verwendung eines statistischen Tests bestätigen die Bedeutung der beobachteten Unterschiede zwischen Protein-Varianten.
- Klicken Sie auf die Registerkarte " Inspektor " und die Statistik -Symbol, um die statistischen Daten anpassen, die für jeden Kanal erforderlich sind.
- Quantifizieren Sie für Protein Immunodetection das rekombinante Protein-Ebene durch Western-Blot mit ImageJ-Software.
- Vor der Analyse Densitometrie wandeln Sie die ursprünglichen raw-Bilder der entwickelten Membran in JPEG-Datei-Format und Graustufen-Modus.
- Stellen Sie Abschnitt Gesetzt Messungen des Menüs " Analyze " zu Grau Mittelwert.
- Klicken Sie auf das Rechteck -Werkzeug von ImageJ und definieren Sie eine Region of Interest (ROI) mit einer einheitlichen Größe für die Band Densitometrie Analyse zu. Zentrieren Sie jedes Band auf der Membran Bild innerhalb des erstellten rechteckigen Rahmens und notieren Sie die Messung eins nach dem anderen mit STRG + M (Maßbefehl).
- Den lokalen Hintergrund neben jedes Band, Anwendung der gleichen Gegend ROI zu messen. Subtrahieren Sie nach der Messung den entsprechenden lokalen Hintergrund aus jedem einzelnen Band.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Dieses Protokoll beschreibt, wie eine Sammlung von 20 Varianten des Peptids Aβ42 zu beschäftigen, wo Phe19 auf alle natürliche proteinogene Aminosäuren28mutiert worden ist. Die theoretische Aggregation Neigungen dieser Proteine können mit zwei verschiedenen bioinformatische Algorithmen (AGGRESCAN und TANGO31,32) analysiert werden. In beiden Fällen macht diese Analyse eine progressive Abstufung der Aggregation Tendenzen, zugeschrieben, als in der Regel die höchsten Werte, hydrophoben Überreste und die niedrigste aufgeladen und polar sind (Abbildung 1 b, 1 C).
Um den intrazellulären Aggregatzustand Aβ42 Varianten experimentell zu verfolgen, erfordert das beschriebene Experiment eine Transformation von einem Plasmid, Codierung für Aβ42 und verschmolzen zu GFP (Abbildung 1A) unter der Kontrolle der Galaktose-induzierbaren GAL1, in S. Cerevisiae. Hefezellen mit dem Ausdruck Aβ42 Varianten nach einer Einarbeitungszeit von 16 h können unter dem Fluoreszenzmikroskop sichtbar gemacht werden. Diese Visualisierung macht es möglich, die Bildung von PI in 10 von 20 Varianten aus der analysierten Sammlung zu bestätigen. Die Anzahl und Größe der fluoreszierenden Aggregate unterscheiden Variante-Variante (Abbildung 2). Abbildung 2A zeigt die PI-Quantifizierung von insgesamt 500 Zellen in jedem der zwei unabhängigen Kulturen. Hier können wir beobachten, ein hervorragendes Abkommen zwischen vorausgesagt und in-Vivo -Aggregation-Eigenschaften.
Um eingegangen, ob die Bildung von Protein-Aggregate in dieser Zelle Modellsystem oxidativen Stress auslösen kann, nutzt dieses Protokoll FC um die zelluläre GFP Fluoreszenz zusammen mit der Fluoreszenz des Prüfpunkts Fluorogenic als Indikator für reaktive Sauerstoff zu überwachen Spezies (ROS) Produktion. Abbildung 3 veranschaulicht die Vorgehensweise und Ergebnisse für Gln und Ile Mutanten, die homogen verteilten und PI-bildenden Aβ42-Varianten entsprechen.
Erstens ist die analysierten Zellpopulation gated (P1) in der FSC-A vs. SSC-A Dot plot, Zelle Detritus aus der Analyse (Abbildung 3A) zu entfernen. Das Fluoreszenzsignal GFP gegen oxidativen Stress Sonde der P1 Bevölkerung in Dot Plot Grafiken wo grüne fluoreszierende Zellen werden in P3 angespritzt vertreten ist (Abb. 3 b). Abbildung 3 und 3D zeigen die Histogramme erstellt um statistische Daten für jede ausdrückliche Variante, einschließlich der CV (Tabelle 2), um den Mittelwert und Median Fluoreszenz der einzelnen fluoreszierenden Marker zu quantifizieren zu erhalten.
Alle Mutanten werden voraussichtlich auf dem gleichen Niveau ausgedrückt werden, da sie in einer einzigen Aminosäure (0,02 % der Aβ42-GFP-Sequenz) unterscheiden. Jedoch kann die proteostase Maschinen auf unterschiedliche Weise auf ihre besondere Aggregation Neigungen reagieren. Daher sind die proteingehalte Ausdruck in Zellextrakten quantifiziert mit einem Aβ-spezifische Antikörper (Abbildung 4). Wir sehen, dass als ein allgemeiner Trend, PI-bildenden Aβ42 Varianten auf einem niedrigeren Niveau als die übrigen in die Zellflüssigkeit homogen verteilt sind.
Wichtige Unterschiede zwischen den Aβ42 Varianten können beobachtet werden, wenn ihre oxidativen Stress Sonde Fluoreszenz, Proteingehalt und GFP Fluoreszenzeigenschaften bezogen auf ihre Fähigkeit, PI und ihre inneren Aggregation Neigungen ( bilden dargestellt werden Abbildung 5). Die Aggregation-anfälliger PI-bildende Varianten entlocken viel niedrigeren oxidativen Stress als Gegenstücke besser löslich (Abbildung 5A, 5 b). Mit diesen Ergebnissen gibt es keine offensichtliche Korrelation zwischen der Größe und der Anzahl der Aggregate pro Einzelzelle (Abbildung 2) und oxidativer Stress. PI-bildende Varianten, vor allem durch Mutation entstehende Variationen mit Trp und Phe an Position 19, befinden sich in der Zelle auf einem niedrigeren Niveau als die homogen verteilt im Zytosol (Abbildung 5, 5D). Die Ausnahme dazu ist der Thr-Mutante, bei denen weniger als 10 % der Zellen PI bilden (Abbildung 1 b, 1 C). Dies entspricht der Tatsache, dass die Aggregation neigende Varianten von der Hefe Qualitätskontrolle Abbau Maschinen28selektiv gelöscht werden. GFP-Fluoreszenz und proteingehalte korrelieren gut für die besser löslichen Aβ42 Formen, aber nicht für die Bildung von PI (Abb. 5E, 5F). Wahrscheinlich deshalb, weil in diesen zellularen Bevölkerungen, die Fluoreszenz aus zwei verschiedenen Kompartimenten, die Zellflüssigkeit und Einschlüsse kommt und ihre relative Beiträge zum gesamten Fluoreszenz zwischen Mutanten34 unterscheiden.

Abbildung 1 . Aggregation Neigung Analyse für die Sammlung von Aβ42-GFP Fusion Konstrukte von der Mutation von Rückständen an Position 19 im Aβ42 Peptid alle natürlichen Aminosäuren abgeleitet. A. dieses Bild zeigt ein 3D-Modell des wt Aβ42 verschmolzen, GFP durch ein Linker (Aβ42, grau; GLP, grüne), basierend auf dem PDB-1EMA für grün fluoreszierendes Protein aus Aequorea Victoria und die 2OTK für die Alzheimer Aβ-Peptid in der Phe19-Seitenkette des wt Aβ42 wird in rot angezeigt. Das Modell ist mit Pymol Software erstellt. Rückstand 19 nacheinander Aβ42 nimmt eine zentrale Position in der zentralen hydrophoben Cluster. Die Balkendiagramme werden mit zwei verschiedenen Bioinformatic Prädiktoren erstellt: B. AGGRESCAN, C. TANGO. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2 . Aβ42-GFP PI-bildende Varianten in S. Cerevisiae Kulturen für 16 h Meinungsäußerung induzierte. A. das Balkendiagramm gibt den Prozentsatz der Zellen, die eine unterschiedliche Anzahl von PI berechnet aus insgesamt 500 fluoreszierenden Zellen für jede Variante in zwei biologischen Wiederholungen enthalten. B. diese Bilder sind repräsentative Fluoreszenz-Mikroskopie Bilder von ausgewählten Aβ42-GFP-Varianten (Phe, Ile, Thr, Trp). Sie wurden unter UV-Licht mit Hilfe eines Anregung Filters für GFP (450-500 nm) und eine Emissionsbereich (515-560 nm) erworben. Die Maßstabsleiste stellt 10 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 3 . Regelung für Flow Cytometry (FC) Analyse of S. Cerevisiae Zellen ausgewählte Aβ42-GFP-Varianten zum Ausdruck zu bringen. A. das Diagramm zeigt gated Hefezellen (P1) in Punkt plottet (FSC-A vs. SSC-A) wo Zelle Detritus aus der Zellpopulation entfernt ist. Die mikroskopischen Aufnahmen der Gln (homogen verteilt) und Ile (PI-Bildung) Varianten sind neben der FC Dot Grundstücke gezeigt. Die Maßstabsleiste stellt 10 µm. B. Diese Scatter Dot Plot Bilder repräsentieren GFP-A vs. oxidativen Stress Sonde in dem eingezäunte Bevölkerung (P3) nur fluoreszierende Zellen enthält ausschließlich das Hintergrundsignal. Die Zelle Frequenz Histogramme sind c. das GFP-Signal (FITC Amplitude) eingezäunt von P1 und D. CellROX (APC Amplitude) eingezäunt von P3. Die Zelle-Übernahme wurde mit einem Durchflusszytometer durchgeführt. Jedes Grundstück stellt 20.000 Veranstaltungen. Q und ich entsprechen bzw. Gln und Ile Aβ42 Mutanten. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4 . Quantifizierung der zellulären Ebenen. Die Abbildung zeigt Western Blots der Gesamt-Proteinfraktionen Aβ42-GFP Mutanten nach 16 h des Ausdrucks in S. Cerevisiae. Die PI-bildende Varianten in grün gefärbt sind und diejenigen in der Zellflüssigkeit diffus verteilt sind hellrot gefärbt. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 5 . Zellulären oxidativen Stress, intrazelluläre GFP Fluoreszenz und zellulären Ebenen Aβ42-GFP Mutanten bestimmt nach 16 h des Ausdrucks in S. Cerevisiae. Die Varianten vertreten auf der x-Achse sind entsprechend ihrer Neigungen vorhergesagten Aggregation von AGGRESCAN (linken) oder TANGO (Rechte Abbildung) bestellt worden. Die PI-bildende Varianten in grün gefärbt sind und die nicht-PI-bildende Varianten sind hellrot gefärbt. A. B. diese Balkendiagramme zeigen den oxidativen Stress Sonde Fluoreszenz Werte durch eine FC-Analyse der Aβ42-GFP Mutanten. C. D. diese Balkendiagramme repräsentieren die proteingehalte, wie durch ein Western-Blot-Densitometrie-Analyse mit ImageJ Software quantifiziert. E. F. diese Balkendiagramme zeigen die GFP-Fluoreszenz-Werte nach einer FC-Analyse. Die Fehlerindikatoren für den oxidativen Stress sondieren und GFP Fluoreszenz Werte darstellen der Koeffizient der Varianz (CV) der FC-gated Zellen. Die Fehlerindikatoren für die proteingehalte Ausdruck repräsentieren ± SE (n = 3). Bitte klicken Sie hier für eine größere Version dieser Figur.
| Kanal | Fluorophor | Erregung Wellenlänge (nm) | Band-Pass-Filter |
| FITC | GFP | 488 | 530/30 |
| APC | CellRox | 635 | 660/20 |
| PerCP | IP | 488 | 585/42 |
Tabelle 1. Laserquelle, Bandpass-Filter und Fluorophore im Durchflusszytometer verwendet.
| GFP-Fluoreszenz | CV | CellROX Fluoreszenz | CV | |
| A | 9316 | 96,4 | 1964 | 127,5 |
| C | 9709 | 91,9 | 1275 | 172,2 |
| D | 11213 | 101,7 | 3443 | 155,8 |
| E | 12256 | 101,1 | 3220 | 152,2 |
| F | 3010 | 96,1 | 1245 | 146.4 |
| G | 11541 | 97,2 | 2947 | 158 |
| H | 7895 | 98,2 | 3582 | 120,8 |
| Ich | 7365 | 97,2 | 1416 | 141,3 |
| K | 10839 | 100.4 | 3102 | 122,1 |
| L | 7605 | 96,9 | 1401 | 161,8 |
| M | 8149 | 96 | 1308 | 170.4 |
| N | 12741 | 97,5 | 3403 | 134,9 |
| P | 9768 | 102,8 | 2629 | 143,6 |
| Q | 13066 | 91,3 | 3354 | 169,9 |
| R | 8537 | 101,3 | 2839 | 127,5 |
| S | 12053 | 99.1 | 3313 | 174,3 |
| T | 10615 | 97,7 | 2213 | 107,7 |
| V | 9169 | 96,1 | 1878 | 121,7 |
| W | 1715 | 94,7 | 1531 | 100 |
| Y | 7574 | 94,5 | 1234 | 138 |
Tabelle 2. Liste der Werte für GFP Fluoreszenz und oxidativem Stress Sonde Fluoreszenz durch die FC-Analyse von Hefezellen mit dem Ausdruck Aβ42-GFP Mutanten für 16 h. Die folgende Tabelle zeigt die mittlere Fluoreszenzintensität (MFI) und der Lebenslauf für jede Mutante.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Eine Vielzahl von Krankheiten ist die Anhäufung von fehlgefaltete Proteine in zellulären Einlagen6,7,8,33verbunden. Wurden viele Anstrengungen unternommen, um die molekularen Mechanismen zu entwirren, die das Auftreten dieser Krankheiten mit rechnerische Ansätze, die nicht ins Konto proteinkonzentrationen nehmen, auslösen oder in-vitro- Ansätze, in denen die Proteinkonzentration während der Reaktion bleibt konstant. Jedoch innerhalb der Zelle sind Proteine ständig synthetisiert und in einer überfüllten und nicht homogene Umgebung abgebaut. Dies erklärt die häufige Abweichungen zwischen in Silico, in Vitro und in Vivo -Aggregation-Eigenschaften der Amyloidogenic Proteine.
Es gibt mehrere Gründe, warum einfache zellmodellen, wie Hefe, sind eine vernünftige Wahl die Aggregation zu studieren und Toxizität von Proteinen, die im Zusammenhang mit neurodegenerativen Erkrankungen in einem mehr biologischen Kontext verbunden. Beispielsweise geben sie uns die Möglichkeit, die Auswirkungen der Proteinaggregation Fehlfaltung auf eine intakte zelluläre Bevölkerung mit schnellen Analysen wie implementiert hier zu analysieren. Allerdings sollten wir berücksichtigen, dass oxidative Ebenen unter Hefestämme unterscheiden können. So sollte zwischen Stämmen oder sogar zwischen verschiedenen Proteinen zu vergleichen, Oxidantien und Reduktionsmitteln wie DVB-t und Diamin, verwendet werden, eine Oxidation/Reduktion Skalierung zu etablieren.
Im Beispiel in diesem Artikel, die bioinformatische Analyse, zelluläre Protein Quantifizierung dargestellt wurden Protein Lokalisierung Bildgebung und gleichzeitige FC Analyse der GFP Aktivität und oxidativen Stress integriert um die molekularen identifizieren Arten für die oxidativen Schäden durch Protein Aggregation Reaktionen hervorgerufen. Die Ergebnisse zeigen, dass die besser löslichen Aβ42 Varianten, anstatt die Aggregation-anfälliger, die höchsten oxidativen Stress fördern. Dies weist auf die diffusiblem Arten gefährlicher Aβ42 Arten in Vivo. Geringere Toxizität der Aggregation Varianten scheint zu reagieren darauf hin, dass diese Konformationen in PI abgesondert werden und zu ihren bevorzugten Abbau von Protein Qualität Maschinen, was zu geringeren Proteingehalt als ihre löslichen Pendants.
Das beschriebene Verfahren beschränkt sich nicht auf die Analyse von den oxidativen Stress durch Aβ42 aggregiert/löslichen Arten produziert und kann auch angewendet werden, um eine Vielzahl von Protein Aggregation Erkrankungen zu untersuchen. Darüber hinaus könnte die Technik nützlich sein, überwachen die Wirkung der Aggregation Inhibitoren auf zellulären oxidativen Stress, so dass diese Moleküle für weitere klinische Anwendungen zu verwerfen, die die Ansammlung von oxidativen aktives Protein Arten zu fördern. Schließlich, solange eine Fluorogenic Sonde zur Verfügung steht, bietet der Ansatz, dass bemerkenswerte Möglichkeiten um die molekularen Spezies verantwortlich für andere toxische Effekte zu identifizieren mit Proteinaggregation in einem sich schnell eine einfache Möglichkeit, die quantitativen Daten verknüpft.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Materials
| Name | Company | Catalog Number | Comments |
| Yeast cells BY4741 | ATCC | 201388 | Genotype: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| pESC(-Ura) plasmid | Agilent Genomics | 217454 | Yeast expression plasmid with a Gal promotor. Selectable marker URA3 |
| Yeast Synthetic Drop-out Medium Supplements | Sigma | Y1501 | Powder |
| Yeast Nitrogen Base Without Amino Acids | Sigma | Y0626 | Powder |
| Raffinose | Sigma | R7630 | Powder |
| Glucose | Sigma | G7021 | Powder |
| Galactose | Sigma | G0750 | Powder |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | Solution 10X |
| CellROX Deep Red Reagent | Life Technologies | C10422 | Free radical cell-permeant fluorescent sensor, non-fluorescent while in a reduced state, and exhibits bright fluorescence upon oxidation by reactive oxygen species (ROS), with absorption/emission maxima at 644/665 nm. |
| Y-PER protein extraction reagent | Thermo Scientific | 78990 | Liquid cell lysis buffer |
| Acrylamide/Bis-acrylamide | Sigma | A6050 | Solution |
| Bradford dye reagent | Bio-Rad | 5000205 | Dye reagent for one-step determination of protein concentration |
| β-amyloid antibody 6E10 | BioLegend | 803001 | Mouse IgG1. The epitope lies within amino acids 3-8 of beta amyloid (EFRHDS). |
| Goat anti-mouse IgG-HRP conjugate | Bio-Rad | 1721011 | |
| Membrane Immobilon-P, PVDF | Millipore | IPVH00010 | |
| Luminata forte | Merk | WBLUF0100 | Premixed, ready to use chemiluminescent HRP detection reagent |
| Phenylmethanesulfonyl fluoride solution (PMSF) | Sigma | 93482 | Protease inhibitor. Dissolved at 0.1 M in ethanol |
| FACSCanto flow cytometer | BD Biosciences | 657338 | Equipped with a 488 nm blue laser for the detection of GFP, and 635 nm red laser / 530/30 nm BP filter and 660/20 BP filter |
| Mini Trans-Blot Electrophoresis Transfer cell | Bio-Rad | 1703930 | Protein transference system |
| Mini-PROTEAN Tetra Handcast Systems | Bio-Rad | 1658000FC | Electrophoresis system |
References
- Frydman, J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annual Review of Biochemistry. 70, 603-647 (2001).
- Hartl, F. U., Bracher, A., Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature. 475, 324-332 (2011).
- Wong, E., et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nature Communications. 3, (2012).
- Winkler, J., Tyedmers, J., Bukau, B., Mogk, A. Chaperone networks in protein disaggregation and prion propagation. Journal of Structural Biology. 179, 152-160 (2012).
- Glickman, M. H., Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological Reviews. 82, 373-428 (2002).
- Dobson, C. M. Getting out of shape. Nature. 418, 729-730 (2002).
- Chiti, F., Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annual Review of Biochemistry. 75, 333-366 (2006).
- Aguzzi, A., O'Connor, T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery. 9, 237-248 (2010).
- Rapezzi, C., et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nature Reviews Cardiology. 7, 398-408 (2010).
- Deas, E., et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxidants & Redox Signaling. 24, 376-391 (2016).
- Brundin, P., Melki, R., Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature Reviews Molecular Cell Biology. 11, 301-307 (2010).
- Frost, B., Diamond, M. I. Prion-like mechanisms in neurodegenerative diseases. Nature Reviews Neuroscience. 11, 155-159 (2009).
- Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431, 805-810 (2004).
- Ross, C. A., Poirier, M. A. Opinion: what is the role of protein aggregation in neurodegeneration? Nature Reviews Molecular Cell Biology. 6, 891-898 (2005).
- Mishra, R., Sjölander, D., Hammarström, P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Molecular BioSystems. 7, 1232-1240 (2011).
- Klunk, W. E., Jacob, R. F., Mason, R. P. Quantifying amyloid by congo red spectral shift assay. Methods in Enzymology. 309, 285-305 (1999).
- Khurana, V., Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nature Reviews Neuroscience. 11, 436-449 (2010).
- Tenreiro, S., Outeiro, T. F. Simple is good: yeast models of neurodegeneration. FEMS Yeast Research. 10, 970-979 (2010).
- Moosavi, B., Mousavi, B., Macreadie, I. G. Yeast model of amyloid-β and Tau aggregation in Alzheimer's disease. Journal of Alzheimer's Disease. 47, 9-16 (2015).
- Tenreiro, S., Munder, M. C., Alberti, S., Outeiro, T. F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. Journal of Neurochemistry. 127, 438-452 (2013).
- Yang, J., Hao, X., Cao, X., Liu, B., Nyström, T. Spatial sequestration and detoxification of huntingtin by the ribosome quality control complex. eLife. 5, (2016).
- Braun, R. J., Büttner, S., Ring, J., Kroemer, G., Madeo, F. Nervous yeast: modeling neurotoxic cell death. Trends in Biochemical Sciences. 35, 135-144 (2010).
- Figley, M. D., Gitler, A. D. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Diseases. 1, e24420 (2013).
- Cooper, A. A., et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 313, 324-328 (2006).
- Johnson, B. S., McCaffery, J. M., Lindquist, S., Gitler, A. D. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 105, 6439-6444 (2008).
- Bharathi, V., et al. Use of ade1 and ade2 mutations for development of a versatile red/white color assay of amyloid-induced oxidative stress in saccharomyces cerevisiae. Yeast. 33, 607-620 (2016).
- Pallarès, I., Ventura, S. Advances in the prediction of protein aggregation propensity. Current Medicinal Chemistry. , (2017).
- Villar-Piqué, A., Ventura, S. Protein aggregation propensity is a crucial determinant of intracellular inclusion formation and quality control degradation. Biochimica et Biophysica Acta. 1833, 2714-2724 (2013).
- Navarro, S., Villar-Piqué, A., Ventura, S. Selection against toxic aggregation-prone protein sequences in bacteria. Biochimica et Biophysica Acta. 1843, 866-874 (2014).
- Morell, M., de Groot, N. S., Vendrell, J., Avilés, F. X., Ventura, S. Linking amyloid protein aggregation and yeast survival. Molecular BioSystems. 7, 1121-1128 (2011).
- Conchillo-Solé, O., et al. AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics. 8, 65 (2007).
- Fernandez-Escamilla, A. M., Rousseau, F., Schymkowitz, J., Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology. 22, 1302-1306 (2004).
- Renner, M., Melki, R. Protein aggregation and prionopathies. Pathologie Biologie.(Paris). 62, 162-168 (2014).
- Carija, A., Navarro, S., de Groot, N. S., Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biology. 12, 699-711 (2017).
