

Summary
Agrégation de la protéine provoque un stress oxydatif cellulaire. Ce protocole décrit une méthode pour surveiller les États intracellulaires des protéines amyloïdogène et le stress oxydatif qui leur sont associés, à l’aide de cytométrie en flux. L’approche est utilisée pour étudier le comportement des variantes solubles et sujette à l’agrégation du peptide amyloïde-β.
Abstract
Repliement et agrégation en conformations amyloïdes ont été associés à l’apparition et la progression de maladies neurodégénératives. Cependant, il y a encore peu d’informations sur les protéines insolubles comment agrégats exercent leurs effets toxiques in vivo. Les organismes de modèle simple de procaryotes et eucaryotes, comme les bactéries et les levures, ont largement contribué à notre compréhension actuelle des mécanismes à l’origine de la formation d’amyloïde intracellulaire, propagation des agrégats et la toxicité. Dans ce protocole, l’utilisation de levure est décrit comme un modèle à disséquer la relation entre la formation d’agrégats de protéines et de leur impact sur le stress oxydatif cellulaire. La méthode associe la détection de l’État soluble/agrégés intracellulaire d’une protéine amyloïdogène la quantification du dommage oxydatif cellulaire résultant de son expression à l’aide de cytométrie en flux (FC). Cette approche est simple, rapide et quantitative. L’étude illustre la technique en mettant en corrélation le stress oxydatif cellulaire provoqué par un grand nombre de variantes de peptide amyloïde-β avec leurs propensions agrégation intrinsèque respectifs.
Introduction
Protéostasie est un déterminant fondamental du processus de remise en forme et le vieillissement cellulaire. Dans les cellules, homéostasie des protéines est maintenue par contrôle de la qualité sophistiqué protéine réseaux ayant pour but d’assurer le repliement correct des conformères les protéines mal repliées par chaperons et/ou leur protéolyse ciblée avec des mécanismes bien conservés plusieurs1 ,2,3,4,5. Un grand nombre d’études soutiennent le lien entre le déclenchement et la progression d’un large éventail de maladies humaines et l’échec de protéostasie, conduisant à un mauvais repliement des protéines et de l’agrégation. Par exemple, la présence de dépôts protéiques est considéré comme une caractéristique pathologique des nombreuses maladies neurodégénératives, comme Alzheimer, Parkinson, Huntington maladies6,7,8, prionogenic maladies et non-dégénératives amyloses9. Il a été suggéré que premières assemblées oligomères et protofibrillar dans la réaction d’agrégation sont les éliciteurs principales de cytotoxicité, établissant des interactions avec d’autres protéines aberrantes dans le milieu cellulaire bondé10. En outre, des inclusions protéiques (PI) peuvent être transmises entre les cellules, se propageant leur effet toxique11,12. Par conséquent, il est possible que la formation de PI pourrait en effet constituer un mécanisme de détoxification qui limite la présence d’espèces agrégées dangereux vers des emplacements spécifiques dans la cellule, où ils peuvent être transformés ou accumulés sans effets secondaires majeurs 13 , 14.
Norme in vitro des approches biochimiques ont fourni des renseignements importants sur les différentes espèces qui peuplent les réactions de l’agrégation et leurs propriétés15,16. Toutefois, les conditions utilisées dans ces essais sont clairement différentes de ceux qui se produisent au sein de la cellule et, par conséquent, la question leur pertinence physiologique. En raison de la conservation remarquable des voies cellulaires tels que le contrôle de la qualité protéique, autophagie ou le règlement de l’oxydo-réduction cellulaire état17,18 chez les eucaryotes19,20,21 ,22,23, la levure Saccharomyces cerevisiae (S. cerevisiae) s’est imposé comme un modèle cellulaire simple privilégié pour étudier les déterminants moléculaires de l’agrégation des protéines et son impact cytotoxique associé dans des environnements biologiquement pertinente24,25,26.
Propension de l’agrégation de protéines est une fonctionnalité intrinsèquement encodée dans la séquence primaire. Ainsi, la formation de structures de type amyloïde peut être prédite basée sur l’identification et l’évaluation de l’activité de promotion à l’agrégation de polypeptides27régions. Cependant, malgré le succès des algorithmes bioinformatiques pour prévoir les propriétés de l’agrégation in vitro de séquences protéiques, elles sont loin de prévoir comment ces penchants se traduiront in vivo des effets cytotoxiques. Les études qui portent sur le lien entre l’état agrégé d’une protéine donnée et ses dommages cellulaires associées de manière systématique peuvent aider à contourner cette limitation computationnelle. Ce sujet est abordé dans la présente étude, en profitant d’un grand nombre de variantes du peptide amyloïde-β Aβ42 ne différant que par un seul résidu, mais affichant une gamme continue de propensions de l’agrégation en vivo28. En particulier, une approche axée sur les FC pour déterminer l’espèce conformationnelle représentent les dommages oxydatifs induits par agrégation sujettes aux protéines dans les cellules de levure est décrite. La méthodologie fournit de nombreux avantages comme la simplicité, la capacité de haut débit et une mesure quantitative précise. Cette approche a permis de confirmer que le jeu PI un rôle protecteur contre le stress oxydatif.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Cultures de S. cerevisiae et Expression de la protéine
Remarque : Les variantes Aβ pièce propensions d’agréger les relative due à une mutation dans un seul résidu à la position 19 (Phe19) du peptide Aβ42 (Figure 1 a). Ces variantes de peptide sont marquées par la protéine fluorescente verte (GFP), constituant ainsi une agrégation journaliste (Figure 1)29.
- Transformer les plasmides codant pour 20 variantes Aβ42-GFP dans des cellules de levure avec une BY4741 fond parental (MATa sa3Δ1 leu2Δ0 a rencontré15Δ0 ura3Δ0)30. Chaque plasmide codant pour un mutant Aβ42 fusionné à la GFP par un éditeur de liens GSSGSSG. Les plasmides sont pESC(-URA) et incluent le marqueur sélectionnable URA329.
- Préparer le milieu synthétique complet sans uracile (SC-URA) avec les composants suivants : 1,9 g/L azote de levure base sans uracile, sulfate d’ammonium 5 g/L et 900 mL de ddH2O combiné avec 100 mL de glucose 20 % (p/v).
- Mettez une colonie les transformé des cellules de levure dans 20 mL de milieu SC-URA contenant 2 % de glucose et poussent les cultures du jour au lendemain à 30 ° C sous agitation à 210 t/mn.
- Le lendemain, poussent les nouvelles cultures de cellules de levure par inoculation 100µl de la culture au jour le jour dans 5 mL de milieu frais de SC-URA.
- Une OD590 de 0,5, centrifuger les cultures à 3 000 x g pendant 4 min, jeter le surnageant et remettre en suspension les cellules dans le même volume de milieu SC-URA frais contenant 2 % raffinose.
- Incuber à 30 ° C sous agitation à 210 tr/min pendant 30 min. Puis, centrifuger les cellules à 3 000 x g pendant 4 min et éliminer le surnageant. Remettre en suspension les cellules dans un milieu frais SC-URA contenant 2 % de galactose pour induire l’expression de protéine recombinante.
- Après 16 h d’induire l’expression de la protéine dans un milieu SC-URA contenant 2 % de galactose, récolter 1 mL des cellules induits dans des microtubes stériles à par centrifugation à 3 000 x g pendant 4 min.
Remarque : Les différences principales se produisent après qu’une période d’induction de 16 h. Induced levures exprimant des variantes Aβ42-GFP peut être visualisée par microscopie à fluorescence pour déterminer la distribution de protéines recombinantes dans les cellules (Figure 2 b).
2. coloration de la cellule
Remarque : Des cellules fraîches non induite sont requis comme témoin négatif de fixer un seuil de fluorescent au cours d’une analyse FC.
- Prenez les cellules de levure induite par h 16 et de déterminer leur densité optique à 590 nm (OD590). Diluez-les dans une saline stérile tamponnée au phosphate (PBS) à une OD590 de 0,1.
- Préparer 1 x solution saline tamponnée au phosphate comme suit : ajouter 8 g de NaCl, 0,2 g de KCl, 1,44 g de Na2HPO4, et de 0,24 g de KH2PO4 à 800 mL de ddH2O. remplir la solution jusqu'à 1 L de ddH2O après ajustement du pH à 7.4 avec HCl filtrer le tampon à travers un filtre de 0,2 µm.
- Transférer les suspensions cellulaires exprimant à tubes correctement identifiés (polystyrène de mm fond rond de 12 x 75) et de les protéger de la lumière. Assurez-vous qu’ils sont prêts à être chargés sur le cytomètre en flux pour l’analyse, ainsi que de cellules non induite et non colorés.
- Ajouter la sonde du stress oxydatif (p. ex., CellROX rouge foncé) pour chaque échantillon à une concentration finale de 5 µM et incuber les cellules à 30 ° C pendant 30 min à l’obscurité.
- Après incubation, laver les cellules 3 fois avec 1 x PBS et les remettre en suspension dans le même volume de tampon avant leur analyse par le FC.
3. analyse en cytométrie en flux
Remarque : Utilisez une configuration FC avec des lasers appropriés et de filtres pour détecter GFP et le signal de fluorescence de sonde le stress oxydatif. Un cytomètre en flux équipé d’un laser à 488 nm bleu pour la détection des GFP et un laser de 635 nm rouge pour la détection de la fluorescence de sonde de stress oxydatif peut être utilisé. Acquisition de fluorescence d’émission de GFP et sonde le stress oxydatif est exécutée avec un filtre de nm BP 530/30 et un filtre de BP 660/20, respectivement (tableau 1).
- Cliquez sur le Nouveau panneau de feuille de calcul ouverte et créer les parcelles de dot acquisition suivants.
- Sélectionnez Scatter Plot outil dans la barre d’outils et de créer un terrain avec les variables secondaires Scatter-zone (SSC-A) sur l’axe des ordonnées vs Forward Scatter-zone (FSC-A) à une échelle linéaire sur l’axe des abscisses.
- Cliquez sur l' Outil de traçage Scatter de la barre d’outils et de créer un terrain avec les variables FL1-zone (FITC) sur l' axe des abscisses vs. LV3-zone (APC) dans une échelle logarithmique sur l’axe y.
- Cliquez sur l’icône Paramètres Instruments . Cliquez sur l’onglet de Compensation et de définir tous les niveaux de rémunération. Cliquez sur l’onglet Acquisition et sélectionnez un nombre total de 20 000 événements à enregistrer.
- Cliquez sur l’icône de Débit pour passer le débit d’eau faible.
- Cliquez sur l’onglet Acquire pour commencer à courir les cellules non induite, colorées ou non et d’ajuster la tension dans les Paramètres de l’Instrument de l’attaquant et la diffusion latérale jusqu'à ce que la population se répartit dans le quadrant de centre-gauche.
- Cliquez sur l’icône de polygone pour définir une zone R1 autour de la population de cellules à l’exclusion des débris cellulaires et utilisez cette population fermée P1 = R1 pour toutes les parcelles dot fluorescent et représentations de l’histogramme (Figure 3).
- Pour ajuster la tension de la PMT du signal de fluorescence, exécuter les cellules non colorés dans la parcelle de dot FL1-LV3, réglage du gain dans l’onglet Configuration de l’Instrument , jusqu'à ce que les cellules sont distribués dans le quadrant inférieur gauche.
- Remplacez l’exemple les cellules induites pour mesurer la fluorescence GFP. Dans l’onglet Configuration de l’Instrument , réglez le gain à la FSC-A vs FL1 (FITC) lorsque la population est répartie dans le quadrant inférieur droit. Définir la population de cellules positives avec une porte (Gate P2).
- L’échantillon à la non induite par le changement colorées des cellules pour afficher le stress oxydatif de la fluorescence sur une parcelle de dot FSC-A vs LV3 (APC). Accordez le gain jusqu'à ce que la population cellulaire est distribuée dans le quadrant supérieur gauche. Porte la population de cellules positives en P3.
- Faire deux parcelles d’histogramme avec l’icône histogramme pour représenter la fluorescence de la cellule. Pour représenter l’intensité de la fluorescence, assurez-vous que FL1 (FITC) représentant la fluorescence GFP est dans l’axe des abscisses, et LV3 (APC) représentant la fluorescence est dans l’axe des ordonnées, appliquant les populations P2 et P3 sur une échelle logarithmique, respectivement.
NOTE : Superpositions histogramme sont une bonne façon d’illustrer les différences entre les profils de fluorescence des cellules exprimant les différentes variantes de bêta-amyloïdes.
4. recombinant Protein immunodétection
- Pour déterminer les niveaux d’expression de protéine, récolter les cultures de levures induites pour 16 h par centrifugation à 4 000 x g pendant 6 min et stocker les pellets de cellule à-80 ° C.
- Remettre en suspension les cellules recueillies exprimant la protéine recombinante pendant 16 h dans du PBS. Préparer des suspensions de cellules 200 µL de chaque mutant Aβ42-GFP à une OD590 de 20.
- Pour l’analyse de fraction de protéine totale, récolter 100 µL de chaque mutant par centrifugation à 14 000 x g pendant 20 min et remettre en suspension les granules cellulaires dans le même volume de tampon de lyse levure Y-par (50 mM Tris-HCl pH 8,0, 1 % DMSO, 200 mM NaCl, EDTA 1 mM contenant 10 mg/mL SB3-14 (myr istyl sulfobétaïne) complétée avec le fluorure phénylméthylsulfonyle (PMSF) de 1 mM).
- Incuber les échantillons pendant 20 min à température ambiante sous agitation légère.
- Déterminer la concentration d’extrait de protéine à l’aide de l’analyse de Bradford. Charger jusqu'à 5 µg d’extrait de protéines de l’échantillon sur un gel de l’électrophorèse SDS-PAGE 15 % d’acrylamide et tache sur une polyvinylidène difluoride (PVDF) membrane à 100 V pour 60 min.
- Préparer un Bradford réactif avec 100 mg de bleu de Coomassie brillant bleu G-250 dissoute dans 50 mL d’éthanol à 95 % et ajouter 100 mL d’acide phosphorique à 85 % (p/v). Puis, lorsque la teinture est complètement dissous, diluer le mélange avec les ddH2O à 1 L et filtrer sur papier filtre cellulose juste avant utilisation.
Remarque : Le réactif de Bradford devrait être brun clair. - Préparer un étalon d’albumine sérique bovine (BSA) allant de 5 à 100 µg de protéine pour 1 000 µL de réactif de Bradford.
- Ajoutez 1-10 µL de protéine extrait à 1 mL de réactif de Bradford et incuber le mélange à température ambiante pendant 5 min.
- Mesurer l’absorbance des normes et les échantillons d’extrait de protéine à OD595.
- Pour l’analyse, faire un complot avec les valeurs d’absorbance obtenues pour les normes vs. µg de protéines. D’après la courbe d’étalonnage, déterminer les concentrations des échantillons originaux de la quantité de protéines, compte tenu du volume et la dilution, le cas échéant.
- Préparer un Bradford réactif avec 100 mg de bleu de Coomassie brillant bleu G-250 dissoute dans 50 mL d’éthanol à 95 % et ajouter 100 mL d’acide phosphorique à 85 % (p/v). Puis, lorsque la teinture est complètement dissous, diluer le mélange avec les ddH2O à 1 L et filtrer sur papier filtre cellulose juste avant utilisation.
- Rincer la membrane avec 1 x Tris salin (SCT), 0,1 % Tween 20 (TTBS) et la bloquer à l’aide de 5 % (p/v) de lait sec dans 1 x TTBS.
- Pour préparer 1 L de 10 x TBS, dissoudre 24 g de base Tris et 88 g de NaCl dans 900 mL d’H2OJJ et ajuster le pH à 7,6. Ajouter ddH2O pour un volume final de 1 L. Pour une solution de 1 x, mélanger 1 partie de stock de x 10 en 9 parties de ddH2O.
- Incuber la membrane pendant 1 h avec un 6E10 dilué 1 : 1 000 anticorps primaire de β-amyloïde et pendant 1 h à température ambiante avec une chèvre de l’anticorps secondaire Conjugué IgG-HRP anti-souris dilué 01:10, 000.
- Développer des membranes en utilisant 1 mL de réactif fluorescent Luminata et quantifier des bandes par densitométrie, comme indiqué au point 5.2.
5. analyse
- Pour analyser les données obtenues par FC, créer une table affichant la moyenne intensité de fluorescence (IFM) et la fluorescence médian avec son écart-type correspondant et/ou le coefficient de variation (CV) pour fluorescence GFP et les niveaux de stress oxydatif.
- Cliquez sur l’onglet de l’inspecteur et l’icône de statistiques pour personnaliser les données statistiques qui sont requises pour chaque canal.
Remarque : Lorsque l'on compare les variants protéiques, convertir les données de valeurs de fluorescence un fond pliable ou la résolution métriques (RD) :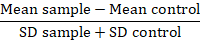
Dans cette mesure, l’échantillon moyen est la fluorescence moyenne des échantillons de problème, le contrôle de la moyens est la moyenne fluorescence des cellules utilisé comme témoin et le SD est l’écart type de la fluorescence de moyenne. Facteur de RD est un indice plus précis que le valeur MFI il calcule non seulement la différence entre les échantillons, mais aussi comptes visant la diffusion des données. Ce calcul est particulièrement pertinent lorsqu’en comparant les données provenant des différentes expériences effectuées au fil du temps. Une fois que les RDs ont été calculées, l’utilisation d’un test statistique est conseillée de confirmer l’importance des différences observées entre les variantes de la protéine.
- Cliquez sur l’onglet de l’inspecteur et l’icône de statistiques pour personnaliser les données statistiques qui sont requises pour chaque canal.
- Pour les protéines immunodétection, quantifier les niveaux de la protéine recombinante par Western blot à l’aide de logiciels ImageJ.
- Avant l’analyse de la densitométrie, convertir les images raw originales de la membrane développée en fichier format et niveaux de gris en mode JPEG.
- Ajuster la section Mesures de la valeur du menu Analyze pour Signifier la valeur de gris.
- Cliquez sur l’outil Rectangle de ImageJ et définir une région d’intérêt (ROI) avec une taille uniforme pour l’analyse de la densitométrie de bande. Centre de chaque bande sur l’image de la membrane à l’intérieur de l’encadrement rectangulaire créée et enregistrer la mesure un par un en utilisant Ctrl + M (la commande de mesure).
- Mesurer le contexte local adjacent à chaque bande, en appliquant la même zone ROI. Après la mesure, soustraire le fond correspondant local de chaque bande.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Ce protocole décrit comment employer une collection de 20 variantes du peptide Aβ42 où Phe19 a été muté à tous les protéinogéniques naturels acides aminés28. Les propensions théorique de l’agrégation de ces protéines peuvent être analysées en utilisant deux algorithmes de bioinformatic différentes (AGGRESCAN et TANGO31,32). Dans les deux cas, cette analyse rend une gradation progressive des tendances de l’agrégation, attribuant, en règle générale, les valeurs les plus élevées de résidus hydrophobes et le plus bas à celles chargées et polaires (Figure 1 b, 1C).
Pour suivre expérimentalement l’état d’agrégation intracellulaire de différentes variantes de Aβ42, l’expérience décrite exige une transformation d’un plasmide codant pour Aβ42 et fusionnée à la GFP (Figure 1 a), sous le contrôle de la galactose-inducible GAL1, dans S. cerevisiae. Les cellules de levure exprimant Aβ42 variantes après une période d’induction de 16 h peuvent être visualisées sous un microscope à fluorescence. Cette visualisation permet de confirmer la formation de PI dans 10 des 20 variantes de la collection analysée. Le nombre et la taille des agrégats fluorescents diffèrent de la variante de la variante (Figure 2). Figure 2 a montre la quantification de PI sur un total de 500 cellules dans chacune des deux cultures indépendantes. Ici, nous pouvons observer un excellent accord entre les deux prédit et in vivo des propriétés agrégation.
Pour déterminer si la formation d’agrégats de protéines peut déclencher le stress oxydatif dans ce système de cellule de modèle, ce protocole utilise des FC pour surveiller la fluorescence GFP cellulaire ainsi que la fluorescence de la sonde fluorogène comme indicateur de réactives de l’oxygène production (DRO). Figure 3 illustre la procédure et a obtenu des résultats pour Gln et Ile des mutants, qui correspondent respectivement aux variantes Aβ42 homogènement distribués et formant des PI.
Tout d’abord, la population de cellules analysées est fermée (P1) dans le point de FSC-A vs de la SSC-A tracer enlever les détritus de la cellule de l’analyse (Figure 3 a). Le signal de fluorescence de GFP contre le stress oxydatif tige de la population de P1 est représenté dans les graphes de terrain point où les cellules fluorescentes vertes sont bloquées en P3 (Figure 3 b). Figure 3 et 3D montrent les histogrammes créées pour recueillir des données statistiques pour chaque variante exprimé, y compris le CV (tableau 2), afin de quantifier la moyenne et la médiane fluorescence de chaque marqueur fluorescent.
Tous les mutants sont censés s’exprimer aux mêmes niveaux, car ils diffèrent par un seul acide aminé (0,02 % de la séquence Aβ42-GFP). Toutefois, la machinerie protéostasie peut répondre de manière différentielle à leurs propensions agrégation particulière. Par conséquent, les niveaux d’expression de protéine dans des extraits cellulaires sont quantifiés à l’aide d’un anticorps spécifique Aβ (Figure 4). Nous voyons que, comme une tendance générale, formant des PI Aβ42 variantes sont présents à des concentrations plus faibles que ceux qui sont restés homogènement distribués dans le cytosol.
Différences importantes entre les variantes Aβ42 peuvent être observées lorsque leur fluorescence de sonde de stress oxydatif, des niveaux de protéines et propriétés de fluorescence GFP sont représentées par rapport à leur capacité à former des PI et leurs propensions intrinsèque de l’agrégation ( Figure 5). L’agrégation plus sujettes formant des PI variantes provoquer le stress oxydatif beaucoup plus faible que leurs homologues plus solubles (Figure 5 a, 5 b). Avec ces résultats, il n’y a pas de corrélation évidente entre la taille et le nombre d’agrégats par cellule individuelle (Figure 2) et des niveaux de stress oxydatif. PI-formant variantes, en particulier les mutants portant Trp et Phe au poste 19, sont présents dans la cellule à des niveaux inférieurs à ceux distribués de façon homogène dans le cytosol (Figure 5, 5D). L’exception à cette règle est le mutant Thr, dont moins de 10 % des cellules forment PI (Figure 1 b, 1C). Cela correspond au fait que les variantes plus sujettes à l’agrégation sont sélectivement effacées par la levure contrôle qualité dégradation machines28. Fluorescence GFP et le taux de protéines sont en corrélation bien pour les formes Aβ42 plus solubles, mais pas pour celui qui a fait PI (Figure 5E, 5F). C’est probablement parce que ces populations cellulaires, la fluorescence provient de deux différents compartiments, le cytosol et les inclusions, et leurs contributions respectives à la fluorescence totale diffèrent entre mutants34.

Figure 1 . Analyse de propension de l’agrégation pour la collecte des constructions de fusion de GFP-Aβ42 dérivée de la mutation du résidu en position 19 du peptide Aβ42 par tous les acides aminés naturels. A. cette image montre un modèle 3D du wt Aβ42 fusionnée à la GFP par un linker (Aβ42, gris ; GFP, vert), basé sur le 1EMA de l’APB pour une protéine fluorescente verte de Aequorea victoria et le 2OTK pour le peptide Aβ Alzheimer dans lequel la chaîne latérale de Phe19 du wt Aβ42 apparaît en rouge. Le modèle est créé à l’aide de logiciels Pymol. Résidu 19 dans la séquence Aβ42 occupe une position centrale dans le complexe central hydrophobe. Les graphiques à barres sont créés avec deux prédicteurs de bioinformatic différents : B. AGGRESCAN, C. TANGO. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2 . Aβ42-GFP PI-formant les variantes dans de S. cerevisiae cultures induites pendant 16 h d’expression. A. le curseur graphique indique le pourcentage de cellules contenant un nombre différent de PI calculée sur un total de 500 cellules fluorescentes pour chaque variante dans deux réplicats biologiques. B. ces images sont des images de microscopie fluorescente représentatifs de certaines variantes Aβ42-GFP (Phe, Trp, Ile, Thr). Ils ont été acquis sous la lumière UV à l’aide d’un filtre d’excitation pour GFP (450-500 nm) et une gamme d’émissions (515-560 nm). La barre d’échelle représente 10 µm. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 3 . Régime d’écoulement cytometry (FC) analyse of S. cerevisiae cellules exprimant certaines variantes Aβ42-GFP. A. le graphique montre des cellules de levure fermée (P1) dans les parcelles de dot (FSC-A vs SSC-A) où les détritus cellulaires est supprimé de la population cellulaire. Les images microscopiques de Gln (distribuée de façon homogène) et variantes de l’Ile (formant des PI) sont affichés à côté de que parcelles de la dot FC. La barre d’échelle représente 10 µm. B. Ces images de terrain scatter points représentent GFP-A vs sonde de stress oxydatif dans lequel la population fermée (P3) comprend des cellules seulement fluorescentes, excluant le signal de fond. Les histogrammes de fréquence cellulaire sont des C. le signal GFP (amplitude FITC) dépendant de P1 et D. CellROX (amplitude de l’APC) dépendant de P3. L’acquisition de la cellule a été réalisée avec un cytomètre en flux. Chaque parcelle représente 20 000 événements. Q et j’ai correspondent à des mutants Gln et Ile Aβ42, respectivement. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 4 . Quantification des niveaux de protéines cellulaires. Cette figure illustre les transferts Western des fractions de protéines totales de mutants Aβ42-GFP après 16 h d’expression chez S. cerevisiae. Les variantes de formatrices de PI sont colorés en vert et ceux diffusément distribués dans le cytosol sont colorés en rouge clair. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 5 . Stress oxydatif cellulaire intracellulaire fluorescence GFP et des niveaux de protéines cellulaires pour des mutants Aβ42-GFP déterminés après 16 h d’expression dans S. cerevisiae. Les variantes représentées sur l’axe des abscisses ont été commandés selon leurs propensions agrégation prédite par TANGO (panneau de droite) ou AGGRESCAN (panneau de gauche). Les variantes de formatrices de PI sont colorés en vert et les variantes non productrices de PI sont colorés en rouge clair. A. B. ces graphiques à barres indiquent les valeurs de fluorescence de sonde de stress oxydatif obtenus par une analyse FC des mutants Aβ42-GFP. C. D. ces graphiques à barres représentent les niveaux de protéines tel que quantifié par une densitométrie immunobuvardage à l’aide de logiciels ImageJ. E. F. ces graphiques à barres indiquent les valeurs de fluorescence de GFP après une analyse FC. Les barres d’erreur pour le stress oxydatif probe et valeurs de fluorescence GFP représentent le coefficient de variation (CV) des cellules FC-dépendants. Les barres d’erreur pour les niveaux d’expression de protéine représentent ± écart type (n = 3). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
| Canal | Fluorophore | Excitation longueur d’onde (nm) | Filtre passe-bande |
| FITC | GFP | 488 | 530/30 |
| APC | CellRox | 635 | 660/20 |
| PerCP | PROPRIÉTÉ INTELLECTUELLE | 488 | 585/42 |
Le tableau 1. Source laser, filtres passe-bande et fluorophores utilisés dans le cytomètre en flux.
| Fluorescence GFP | CV | Fluorescence CellROX | CV | |
| A | 9316 | 96,4 | 1964 | 127,5 |
| C | 9709 | 91,9 | 1275 | 172,2 |
| D | 11213 | 101,7 | 3443 | 155,8 |
| E | 12256 | 101.1 | 3220 | 152,2 |
| F | 3010 | 96.1 | 1245 | 146,4 |
| G | 11541 | 97,2 | 2947 | 158 |
| H | 7895 | 98,2 | 3582 | 120,8 |
| J’ai | 7365 | 97,2 | 1416 | 141,3 |
| K | 10839 | 100.4 | 3102 | 122.1 |
| L | 7605 | 96,9 | 1401 | 161,8 |
| M | 8149 | 96 | 1308 | 170,4 |
| N | 12741 | 97,5 | 3403 | 134,9 |
| P | 9768 | 102,8 | 2629 | 143,6 |
| Q | 13066 | 91.3 | 3354 | 169,9 |
| R | 8537 | 101.3 | 2839 | 127,5 |
| S | 12053 | 99.1 | 3313 | 174,3 |
| T | 10615 | 97,7 | 2213 | 107,7 |
| V | 9169 | 96.1 | 1878 | 121,7 |
| W | 1715 | 94,7 | 1531 | 100 |
| Y | 7574 | 94,5 | 1234 | 138 |
Le tableau 2. Liste des valeurs pour la fluorescence GFP et de fluorescence de sonde de stress oxydatif obtenue par analyse FC des levures exprimant des mutants Aβ42-GFP pour 16 h. Ce tableau montre l’intensité de fluorescence moyenne (IFM) et le CV pour chaque mutant.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Un large éventail de maladies est lié à l’accumulation de protéines mal repliées en dépôts cellulaire6,7,8,33. De nombreux efforts ont été faits à élucider les mécanismes moléculaires qui déclenchent l’apparition de ces maladies en utilisant des approches informatiques, qui ne tiennent pas les concentrations de protéine compte, ou in vitro s’approche, où la concentration de protéine reste constante au cours de la réaction. Toutefois, au sein de la cellule, protéines sont constamment synthétisées et dégradées dans un environnement encombré et non homogène. C’est ce qui explique les écarts fréquents entre in silico, in vitro et in vivo des propriétés l’agrégation des protéines amyloïdogène.
Il y a plusieurs raisons pour lesquelles les modèles cellulaires simples, telles que la levure, sont un choix raisonnable d’étudier l’agrégation et associés à la toxicité des protéines liées à des maladies neurodégénératives dans un contexte plus biologique. Par exemple, ils nous donnent la possibilité d’analyser l’impact de l’agrégation des protéines pourrait sur une population cellulaire intacte à l’aide d’analyses rapides tel que celui mis en place ici. Cependant, nous devrions considérer que niveaux oxydatifs peuvent différer entre les souches de levures. Par conséquent, pour comparer entre les souches, ou même entre différentes protéines, oxydants et réducteurs, comme le DTT et diamine, devraient servir à fixer un barème d’oxydo-réduction.
Dans l’exemple illustré dans cet article, l’analyse bioinformatique, la quantification des protéines cellulaires, protéines localisation imagerie et analyse simultanée de FC de l’activité de la GFP et des niveaux de stress oxydatif ont été intégrés afin d’identifier le moléculaire espèces responsables pour les dommages oxydatifs induits par les réactions de l’agrégation de protéines. Les résultats démontrent que les variantes Aβ42 plus solubles, plutôt que le plus agrégation sujettes, promouvoir le stress oxydatif plus élevé. Cela souligne l’espèce diffusible étant le plus dangereux des espèces Aβ42 in vivo. La basse toxicité d’agrégation de variantes semble réagir aussi bien sur le fait que ces conformations sont séquestrées dans le PI et à leur dégradation préférentielle par les mécanismes de qualité de protéine, aboutissant à des niveaux inférieurs de protéines que celle de leurs soluble homologues.
La méthode décrite n’est pas limitée à l’analyse de stress oxydant produit par Aβ42 espèces regroupées/soluble et peut également être appliquée à l’étude une variété de troubles de l’agrégation de protéines. En outre, la technique peut être utile pour surveiller les effets des inhibiteurs de l’agrégation sur le stress oxydatif cellulaire, permettant à jeter davantage les applications cliniques de ces molécules qui favorisent l’accumulation des espèces oxydant protéine active. Enfin, tant qu’une sonde fluorogène est disponible, l’approche offre remarquables possibilités d’identifier les espèces moléculaires responsables des autres effets toxiques liée à l’agrégation des protéines dans un rapide un moyen facile, fournissant des données quantitatives.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Materials
| Name | Company | Catalog Number | Comments |
| Yeast cells BY4741 | ATCC | 201388 | Genotype: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| pESC(-Ura) plasmid | Agilent Genomics | 217454 | Yeast expression plasmid with a Gal promotor. Selectable marker URA3 |
| Yeast Synthetic Drop-out Medium Supplements | Sigma | Y1501 | Powder |
| Yeast Nitrogen Base Without Amino Acids | Sigma | Y0626 | Powder |
| Raffinose | Sigma | R7630 | Powder |
| Glucose | Sigma | G7021 | Powder |
| Galactose | Sigma | G0750 | Powder |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | Solution 10X |
| CellROX Deep Red Reagent | Life Technologies | C10422 | Free radical cell-permeant fluorescent sensor, non-fluorescent while in a reduced state, and exhibits bright fluorescence upon oxidation by reactive oxygen species (ROS), with absorption/emission maxima at 644/665 nm. |
| Y-PER protein extraction reagent | Thermo Scientific | 78990 | Liquid cell lysis buffer |
| Acrylamide/Bis-acrylamide | Sigma | A6050 | Solution |
| Bradford dye reagent | Bio-Rad | 5000205 | Dye reagent for one-step determination of protein concentration |
| β-amyloid antibody 6E10 | BioLegend | 803001 | Mouse IgG1. The epitope lies within amino acids 3-8 of beta amyloid (EFRHDS). |
| Goat anti-mouse IgG-HRP conjugate | Bio-Rad | 1721011 | |
| Membrane Immobilon-P, PVDF | Millipore | IPVH00010 | |
| Luminata forte | Merk | WBLUF0100 | Premixed, ready to use chemiluminescent HRP detection reagent |
| Phenylmethanesulfonyl fluoride solution (PMSF) | Sigma | 93482 | Protease inhibitor. Dissolved at 0.1 M in ethanol |
| FACSCanto flow cytometer | BD Biosciences | 657338 | Equipped with a 488 nm blue laser for the detection of GFP, and 635 nm red laser / 530/30 nm BP filter and 660/20 BP filter |
| Mini Trans-Blot Electrophoresis Transfer cell | Bio-Rad | 1703930 | Protein transference system |
| Mini-PROTEAN Tetra Handcast Systems | Bio-Rad | 1658000FC | Electrophoresis system |
References
- Frydman, J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annual Review of Biochemistry. 70, 603-647 (2001).
- Hartl, F. U., Bracher, A., Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature. 475, 324-332 (2011).
- Wong, E., et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nature Communications. 3, (2012).
- Winkler, J., Tyedmers, J., Bukau, B., Mogk, A. Chaperone networks in protein disaggregation and prion propagation. Journal of Structural Biology. 179, 152-160 (2012).
- Glickman, M. H., Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological Reviews. 82, 373-428 (2002).
- Dobson, C. M. Getting out of shape. Nature. 418, 729-730 (2002).
- Chiti, F., Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annual Review of Biochemistry. 75, 333-366 (2006).
- Aguzzi, A., O'Connor, T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery. 9, 237-248 (2010).
- Rapezzi, C., et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nature Reviews Cardiology. 7, 398-408 (2010).
- Deas, E., et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxidants & Redox Signaling. 24, 376-391 (2016).
- Brundin, P., Melki, R., Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature Reviews Molecular Cell Biology. 11, 301-307 (2010).
- Frost, B., Diamond, M. I. Prion-like mechanisms in neurodegenerative diseases. Nature Reviews Neuroscience. 11, 155-159 (2009).
- Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431, 805-810 (2004).
- Ross, C. A., Poirier, M. A. Opinion: what is the role of protein aggregation in neurodegeneration? Nature Reviews Molecular Cell Biology. 6, 891-898 (2005).
- Mishra, R., Sjölander, D., Hammarström, P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Molecular BioSystems. 7, 1232-1240 (2011).
- Klunk, W. E., Jacob, R. F., Mason, R. P. Quantifying amyloid by congo red spectral shift assay. Methods in Enzymology. 309, 285-305 (1999).
- Khurana, V., Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nature Reviews Neuroscience. 11, 436-449 (2010).
- Tenreiro, S., Outeiro, T. F. Simple is good: yeast models of neurodegeneration. FEMS Yeast Research. 10, 970-979 (2010).
- Moosavi, B., Mousavi, B., Macreadie, I. G. Yeast model of amyloid-β and Tau aggregation in Alzheimer's disease. Journal of Alzheimer's Disease. 47, 9-16 (2015).
- Tenreiro, S., Munder, M. C., Alberti, S., Outeiro, T. F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. Journal of Neurochemistry. 127, 438-452 (2013).
- Yang, J., Hao, X., Cao, X., Liu, B., Nyström, T. Spatial sequestration and detoxification of huntingtin by the ribosome quality control complex. eLife. 5, (2016).
- Braun, R. J., Büttner, S., Ring, J., Kroemer, G., Madeo, F. Nervous yeast: modeling neurotoxic cell death. Trends in Biochemical Sciences. 35, 135-144 (2010).
- Figley, M. D., Gitler, A. D. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Diseases. 1, e24420 (2013).
- Cooper, A. A., et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 313, 324-328 (2006).
- Johnson, B. S., McCaffery, J. M., Lindquist, S., Gitler, A. D. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 105, 6439-6444 (2008).
- Bharathi, V., et al. Use of ade1 and ade2 mutations for development of a versatile red/white color assay of amyloid-induced oxidative stress in saccharomyces cerevisiae. Yeast. 33, 607-620 (2016).
- Pallarès, I., Ventura, S. Advances in the prediction of protein aggregation propensity. Current Medicinal Chemistry. , (2017).
- Villar-Piqué, A., Ventura, S. Protein aggregation propensity is a crucial determinant of intracellular inclusion formation and quality control degradation. Biochimica et Biophysica Acta. 1833, 2714-2724 (2013).
- Navarro, S., Villar-Piqué, A., Ventura, S. Selection against toxic aggregation-prone protein sequences in bacteria. Biochimica et Biophysica Acta. 1843, 866-874 (2014).
- Morell, M., de Groot, N. S., Vendrell, J., Avilés, F. X., Ventura, S. Linking amyloid protein aggregation and yeast survival. Molecular BioSystems. 7, 1121-1128 (2011).
- Conchillo-Solé, O., et al. AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics. 8, 65 (2007).
- Fernandez-Escamilla, A. M., Rousseau, F., Schymkowitz, J., Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology. 22, 1302-1306 (2004).
- Renner, M., Melki, R. Protein aggregation and prionopathies. Pathologie Biologie.(Paris). 62, 162-168 (2014).
- Carija, A., Navarro, S., de Groot, N. S., Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biology. 12, 699-711 (2017).
