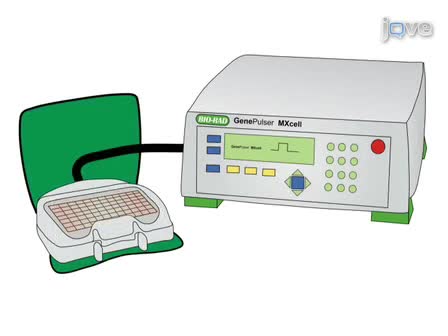
Here we present two techniques for manipulating gene expression in murine retinal ganglion cells (RGCs) by in utero and ex vivo electroporation. These techniques enable one to examine how alterations in gene expression affect RGC development, axon guidance, and functional properties.