

Summary
ここでは、サンプルの限られた量を扱う場合は特に、マイクロRNA(miRNA)の発現レベルのコストと時間効果のハイスループットスクリーニングのツールとしてのマイクロ流体プラットフォームとの組み合わせで最適化された多重逆転写定量PCR(定量RT - PCR)プロトコルについて説明します。
Abstract
開発、組織homoeostasisと疾患の基礎となる重要なプロセスにおけるmiRNAの広範な関与は、研究や医薬品のコミュニティの間で高騰の関心につながっている。 miRNAを研究するために、それは、miRNAレベルの定量化が正確かつ堅牢であることが不可欠です。小さなRNA欠損マウス胚性幹細胞(MESC)に野生型と比較することによって、我々は以前公開されたマルチプレックス定量RT - PCR技術の精度と堅牢性の欠如を明らかにした。ここで、我々は劇的に技術の精度と堅牢性を高めるsingleplexリアルタイム検出、前に前の多重ステップからの過剰なプライマーを離れて精製することを含めて最適化された方法を、説明します。ナノリットルボリュームでマイクロ流体チップ上のテクニックを実行する方法さらに、我々は説明大幅に試薬コストを削減し、時間の効果的なハイスループットなmiRNA発現プロファイリングを可能にする。
Protocol
1。 RNAの抽出:細胞単層培養
(TRIZOLを使用して、製造元のプロトコールに従う。)
| 試薬の名前 | 会社 | #/猫を突く |
| TRIZOLソリューション | インビトロジェン | 15596-018 |
| イソプロピルアルコール | シグマアルドリッチ | 1907年から1964年 |
| クロロホルム | シグマアルドリッチ | C 2432 |
| エタノール | ||
| RNaseフリーの水 |
均質化
- 周りの渦巻きトリゾールを、10 cm 2と皿の面積当たり1mlトリゾールソリューションを追加することで直接培養皿で細胞をホモジナイズし、プレートの完全なカバレッジを確保するために上下にピペッティングし。
相分離
- 室温で5分間均質化したサンプルをインキュベートする。
- 、標識されたチューブに移し、1ミリリットルTrizolSolutionあたりのクロロホルム0.2 mlを加え15秒間手で強くチューブを振る。
- 3分間室温でインキュベートする。
- 2で15分間12,000 xgでサンプルを遠心℃に遠心分離後、混合物は、低赤相、中間相およびRNAを含む無色の上部の水相、に分離し、合計トリゾール溶液体積の60%にする必要があります。
RNA降水量
- RNAは、新鮮な、ラベルの付いたチューブにフェーズを含む、水溶液を移す。
- トリゾール溶液の1mlあたりイソプロピルアルコール0.5 mlを加えることによりRNAを沈殿させる。
- 室温で10分間サンプルをインキュベートする。
- 2で10分間12,000 xgで遠心する℃、
RNA -ウォッシュ
- 遠心分離に続いて、チューブから上清を捨てる。
- トリゾールソリューション、渦の1ml当たりの75%エタノールの少なくとも1 mlを加えることによりRNAのペレットを(チューブの側面と底面に)洗浄する。
- 2で5分間7500 × gで遠心℃、
RNA溶出
- 上清を捨て、1分間再遠心する。過剰な液体を取り除く。
- 5〜10分間空気乾燥ペレット。
注:乾燥RNA上には可溶化することは困難です。 - 上下ソリューションをピペッティングによりRNaseを含まない水でペレットを再溶解し、55℃で10分間インキュベート℃に
- 光度計™分光光度計を用いてRNAの濃度(A260/280比)を測定します。
- -80℃で保存使用直前まで、C。
2。 RNAの抽出:血清
(Mitchellらによって修正された後mirVana PARIS Kitの製造業者のプロトコール1)
| 試薬の名前 | 会社 | #/猫を突く | コメント |
| ミールヴァナPARISキット 2X変性溶液 酸性フェノール:クロロホルム miRNAの洗浄溶液1 miRNAの洗浄液2月3日 | アンビオン | AM1556 | 改良プロトコール |
| 2 - メルカプトエタノール | シグマアルドリッチ | M7522 | |
| エタノール | |||
| DEPCは水を扱わ |
化学物質の調製:
- 2X変性溶液に2 - メルカプトエタノール375μlを加え、よく混ぜる。
- miRNAの洗浄溶液1に100%エタノール21 mlを加え。
- miRNAの洗浄溶液2 / 3に100%エタノール40mlを追加。
サンプル調製:
- 氷上で血清サンプルを解凍。
均質化
- 2X変性溶液の等量(300μl)を(室温にあるはず)と300μlのサンプルを混合し、ボルテックス。
- 氷上で5分間混合物をインキュベートする。
- 酸性フェノールの量を追加:サンプルのライセートに加え、2X変性溶液(600μl)の合計量にクロロホルム等しい。
重要:クロロホルム:酸性フェノールの上にある水性緩衝液を使用しないでください。 - 60秒間ボルテックス混合物。
- 最大速度(> 10,000 × g)で15分間室温で遠心する。遠心分離後、混合物は、上部の水相、中間相および下層の有機相に分離する。
- aqueを削除します。上のOU層(300でなければなりません - いかなるproteinacious"かすんだ"層を含む500μlの)。
- 再びクロロホルムを追加して以前のように水層を再抽出する。
重要:2回目の抽出の水層は(250μl)を体積で日常的に小さくなります。回収されたボリュームに注意してください。
最終的なRNAアイソレーション(全RNA)の調製
- プリヒート(95℃)ヌクレアーゼフリー水。
- 100%エタノールで室温にする必要があります。
最終的なRNAアイソレーション(トータルRNA)
- 回収した水相(例えば300μlを回収した場合、375μlのエタノールを加える)に100%エタノールの1.25ボリュームを加えてよく混ぜる。
- 各サンプルのため、コレクションチューブのいずれかにフィルターカートリッジを置きます。
- フィルターにライセートをピペットで。
- 遠心(10,000 × g)で30秒間、、フロースルーを捨てる30秒間再遠心し、同じコレクションチューブにフィルターカートリッジを交換してください。
RNA -ウォッシュ
- 、15秒のためのフィルターカートリッジと遠心に700μlのmiRNAの洗浄液1を適用するコレクションチューブからフロースルー廃棄し、そして同じコレクションチューブにフィルターカートリッジを交換してください。
- 、500μlのmiRNAのWash Solutionを2 / 3、15秒間遠心するを適用するフロースルーを捨てて、Wash Solutionを2 / 3の第二500μlと繰り返す。
- 1〜2分のためにフロースルーとドライスピンを捨てる。
- 新鮮なコレクションチューブにフィルターカートリッジを置き、100μlのRNaseフリー水(95℃)、2分間近くにキャップとインキュベートを適用する。
- RNAを回収するために〜2分間遠心する。
- -80℃で保存、使用するまで。
3。 RNA -溶液の濃度(5倍)
(製造業者のプロトコールに従って)
| 試薬の名前 | 会社 | #/猫を突く |
| マイクロ遠心フィルターデバイス、Ultracell YM - 3 | ミリポア | 42404 |
| DEPCは水を扱わ |
デバイス:マイクロコンUltracell YM - 3、カットオフSSとDSのヌクレオチド:10
ボリューム:再濃縮RNA溶液の所望の体積は、実験計画に依存しており、対照実験を含める必要があります。
- 貯水池にバイアルおよびピペット溶液にマイクロコンのサンプルリザーバーを挿入します。
メモ:メンブレンに触れないでください。 - 4℃で185分間遠心度× gで最大14000とCを
注:ローターに向かって亀裂帯とポジションバイアル。 - 貯水池の上にRNaseフリーの水のピペット必要なボリューム(例えば、20μl)を、場所のタンクは4で3分間、新しいバイアルとスピンに反転° C、最大3000 x gである
- -80℃で保存、使用するまで。
4。逆転写
(Tangらによってプロトコルに従う。2修正で。)
| 試薬の名前 | 会社 | #/猫を突く | コメント |
| 高容量のcDNAアーカイブキット 10倍のcDNAをアーカイブバッファ のdNTP(100 mM)を モロニーマウス白血病ウイルス(MMLV) 転写酵素(50 U /μl)を逆 | アプライドバイオシステムズ | 4368814 | 改良プロトコール |
| RNaseOUT(40 U /μL) | インビトロジェン | 10777019 | |
| Xプレックスの逆ステムループプライマーミックス(40 nM)を* | 統合されたDNAの技術 | カスタム | シーケンス* |
| DEPCは水を扱わ |
反応液量:5.5μL
注意:ステムループプライマーの終濃度が1〜5 nMの(ここでは2 nM)のでなければならない。
次のようにマスターミックスを(反応あたりのボリューム)を準備します。
| 1。 DEPC -水 | 1.959μlの |
| 2。 10倍RT -バッファ | 0.55μL |
| 3。 RNase阻害剤(40 U /μL) | 0.0715μL |
| 4。のdNTP(100 mM)を | 0.275μlの |
| 5。 Xプレックスの逆ステムループプライマーミックス(40 nM)を* | 0.275μlの |
| 6。 MMLV - RT(50 U /μL) | 0.369μlの |
- マイルxと遠心簡潔に。
- 2μlのサンプルを3.5μlのマスターミックスを追加。
- 以下のRT反応を行います。
16℃20℃60サイクルで30分間、30秒間° C、42℃30秒、50℃1秒、最後に85 C ° MMLV - RTを不活性化するために5分間、4℃∞ 。 - 使用するまで-20℃で保存する。
逆ステムループプライマーの*リストは、次のURLで参照することができますhttp://urology.ucsf.edu/blellochlab/protocols.htm
5。プリアンプ(プリPCR)
(Tangらによってプロトコルに従う。2修正で。)
| 試薬の名前 | 会社 | #/猫を突く | コメント |
| のdNTP(100 mM)を | アプライドバイオシステムズ | 4368814 | ハイキャップ。 cDNAのアーカイブキット。 |
| NoAmpErase UNGと2倍のユニバーサルPCRマスターミックス | アプライドバイオシステムズ | 4324018 | |
| X -プレックスがフォワードプライマーミックス(450 nM)を* | 統合されたDNAの技術 | カスタム | シーケンス* |
| ユニバーサルリバースプライマー(100μM) | 統合されたDNAの技術 | カスタム | シーケンス2 |
| AmpliTaqGoldポリメラーゼ(5 U /μL) | アプライドバイオシステムズ | 4311806 | |
| のMgCl 2(100 mM)を | |||
| DEPCは水を扱わ |
反応容量:27.5μl(22μlMM +5.5μlRT -製品)
注:プリアンプは、RNAの初期濃度が限られている場合は特に、信号強度を高めることが必要である。入力RNAのレベルはPre - PCRサイクルの最適数を決定します。同じ相対的な効率を考えると、各PCRサイクルは、最終製品の濃度を倍増する必要があります。いくつかのmiRNAがより高度に発現されるので、サイクリングを介して避ける。しかし、下の増幅は、特に低レベルで発現のマイクロRNAのため、検出感度が失われる可能性があります。私たちの手でトータルRNAの100ngを使用して12プレ増幅サイクルは、、十分見えた。出発原料、検出可能なmiRNAレベルでの12サイクルの結果が、15サイクルが優れているように見えたとして血清を使用する。最後に、3卵母細胞(卵母細胞3当たり約400 pgのトータルRNA)、16のプリ増幅サイクルで始まるを正常にマイクロRNAの発現レベル4の画面することができました。
フォワードプライマーの*リストは、次のURLで参照することができますhttp://urology.ucsf.edu/blellochlab/protocols.htm
次のようにマスターミックスを(反応あたりのボリューム)を準備します。
| 1。 2倍ユニバーサルMM | 13.75μL |
| 2。 DEPC -水 | 0.792μlの |
| 3。のMgCl 2(100 mM)を | 0.55μL |
| 4。のdNTP(100 mM)を | 1.1μL |
| 5。 X -プレックスがフォワードプライマーミックス(450 nM)の | 3.06μL |
| 6。ユニバーサルRP(100μM) | 1.375μlの |
| 7。 AmpliTaqGoldポリメラーゼ(5 U /μL) | 1.375μlの |
- 混和後スピンダウンする。
- 各5.5μlのRT -製品〜22μlのマスターミックスを追加。
- 以下のプレPCR反応を実行します。
10分間95℃、55℃2分間、10が続きます - 1秒および65のために95℃の18サイクル℃で1分間、4℃∞。
6。プレPCRプロダクトの精製
10パーセントネイティブポリアクリルアミドゲル上でサイズ選択による精製
| 試薬の名前 | 会社 | #/猫を突く |
| 40%アクリルアミド/ビスソリューション | Bio - Rad社 | 161-0144 |
| TEMED | Bio - Rad社 | 161-0801 |
| 10bpのDNAラダー | インビトロジェン | 10821-015 |
| SYBRゴールド核酸ゲル染色 DMSOに10,000 X濃縮 | インビトロジェン | S11494 |
| グリコーゲン(20 mg / mlの) | ロッシュ | 10 901 393 001 |
| バッファ- EB | ||
| 10%のAmmoniumpersulfate(APS) | ||
| TEN -バッファ | ||
| 6XオレンジG | ||
| DEPCは水を扱わ | ||
| ddH2O |
A.ゲルの準備
10%ポリアクリルアミド混合10mlのを作るに追加
| 1。のddH 2 O | 6.5ミリリットル |
| 2。 10倍TBE | 1ミリリットル |
| 3。ポリアクリルアミド(40%) | 2.5ミリリットル |
| 4。 APS(10%) | 80μlの |
| 5。 TEMED | 4μlの |
- DNAラダーを準備します。
- 0.5μlの10bpのDNAラダー
- 4.5μlのBuffer(EB)
- 1μlの6XオレンジG
- それぞれ27.5μlのプレPCR産物は5.5μlを6倍オレンジGを追加。
B.ジェルを実行します。
- 55〜60分間、150Vでゲルを実行します。
注:状況インジケーターとしてブロモフェノールブルーは、20bpの領域で実行されます。
C.ゲルを染色
- シェーカー上で25分間、SYBR - Goldでゲルを染色する。
注 :SYBR - Goldは、光に敏感です。
D.ジェルをカット
- 60から80塩基対からプレPCR産物のバンドを切り出し、ラベルの付いたチューブに移す。
注:低いRNAインプットプレPCR産物のバンドが表示されない場合があります。 - ゲルのバンド(例えばチューブの遠心でチューブを)押しつぶす。
E.再抽出したcDNA
- 300μlを加え37℃TEN -バッファとインキュベートしますローテーター上で4時間。
- 、ラベルの付いたコレクションチューブに液体を移しゲル含有チューブに別の300μlのTEN -バッファを追加し、ローテーター上℃で一晩4℃ですべてをインキュベートする。
- 以前のようにラベルの付いたコレクションチューブに残った液を、転送を吸引除去し、総回収量に注意してください。
F.エタノール沈殿
- 室温の100%エタノールを3倍量を追加。
- 0.5μlのグリコーゲン(20 mg / ml)を追加。
- 渦。
- 少なくとも2時間または℃で一晩-80℃ドライアイスでインキュベートする。
- ペレットのcDNAへの30分間、4℃で遠心機でスピン。
- 流体をダンプ、700μlの室温10分間75%エタノールとスピンを追加します。
- 流体をダンプし、5分間再びスピン。
- 10分間ペレットと空気乾燥を乱すことなく上澄みを吸う。
- きれいな水でペレットを溶解する。
注:集中cDNA溶液の所望の体積は、実験計画に依存して対照実験を含める必要があります。
7。プレPCRプロダクトの精製
スピンカラムに続くプライマーの酵素消化による精製(プロトコール製造に続く)
| 試薬の名前 | 会社 | #/猫を突く |
| ExoSAP - IT | USB -アフィメトリクス | 78250 |
| アプライPCR精製キット | キアゲン | 28004 |
注:私たちの手では排他的な酵素のクリーンアップが正常に削除プライマーはまた、温度は℃の不活性化ステップの間に80まで上昇すると短い製品の変性の可能性に起因する部分、プレ増幅産物の部分的な劣化につながったが酵素。熱不活化を回避し、直接スピンカラムを用いて酵素反応からのPCR産物を精製すると、製品の劣化なしにプライマーを削除します。
- 2μlのExoSAP - ITと5μlのPCR後の製品を混在させること。
- 37℃の残りのプライマーとヌクレオチドを分解する15分間。
- PCR反応および混合の1ボリュームにバッファPBの5ボリュームを追加し、pH指示薬、私はPBをバッファに追加されている場合、混合物の色が黄色であることを確認してください。
- 適切なラックに付属2 mlのコレクションチューブにMinEluteカラムを置きます。
- 1分間MinEluteカラムや遠心するサンプルを適用します。
- フロースルー廃棄し、同じチューブに戻しMinEluteカラムを置き、1分間洗浄し、遠心分離にMinEluteカラムに750μlのBuffer PEを。
- フロースルーを捨て、同じチューブにMinEluteカラムの後ろに置きます。最高速度でさらに1分間カラムを遠心する。
- 1.5 mlのマイクロ遠心チューブにMinEluteカラムを置きます。
- DNAを溶出するために、CENし、メンブレンの中央に10μlのDEPC処理水を追加する列が1分間放置し、1分間trifuge。
8。 Fludigim 96.96定量RT - PCRプロファイリング
| 試薬の名前 | 会社 | #/猫を突く | コメント |
| フリューダイム96.96ダイナミックアレイキット 96.96動的配列 96.96制御ラインの流体 20倍ローディング試薬 | フリューダイム株式会社 | ||
| のミックス ユニバーサルリバースプライマー(1μM) フォワードプライマー(1μM) TaqManプローブ(0.2μM) | 統合されたDNAの技術 | カスタム | シーケンス(1) |
| 2倍のユニバーサルPCRマスターミックス NoAmpErase UNGによる | アプライドバイオシステムズ | 4324018 | |
| トゥイーン |
1。プライミング96.96ダイナミックアレイIFCの
- チップ上のアキュムレータの各々にコントロールラインの液(150μl)を注入する。
- 場所は、IFC(集積流体回路)コントローラにチップとチップに主要な制御線の流体に総理(136x)スクリプトを実行します。
注:チップは、パッケージを開封後24時間を使用する必要がある。チップ上または吸気口の制御線の流体チップが使用できなくなるため、コントロールラインの液をこぼさないように確認してください。チップは、プライミングの60分以内にロードする必要があります。
2。サンプルの準備
2.1前のサンプル調製
使い捨てのプラスチック製のチューブに、サンプルプレミックス(入口あたりのボリューム)を作成するには、以下の表に示すコンポーネントを組み合わせる。
| 2倍のTaqManユニバーサルマスターミックス | 2.5μlの |
| 20倍ローディング試薬 | 0.25μL |
注:ピペットのための超過は、損失を分配するように最終的なボリュームは、2.75μLです
2.2サンプルの調製
よく事前にサンプルミックス(入口あたりのボリューム)で、各サンプルをフォーマット。96に結合する
| プレサンプルミックス | 2.75μL |
| サンプル | 2.25μL |
注:渦と流体を集めるために一時的に96ウェルプレートを遠心する。
入口を5μlにつき、最終的なボリュームは、わずかに高いボリュームを準備することによってアカウントにピペッティング損失を取る。
13倍アッセイの準備2.3
- 13倍アッセイを準備するために96ウェルプレートを使用してください。以下の表に1倍濃度は、13倍のためにスケールアップ。
| ユニバーサルリバースプライマー | 1μmの(1X) | 2のシーケンス |
| フォワードプライマー* | 1μmの(1X) | * |
| 遺伝子特異的なTaqManプローブ# | 0.2μmの | # |
フォワードプライマーの*リストは、次のURLで参照することができますhttp://urology.ucsf.edu/blellochlab/protocols.htm
#TaqManプローブのリストは、次のURLで参照することができますhttp://urology.ucsf.edu/blellochlab/protocols.htm
- 0.25パーセント、最終のTweenの濃度をTweenで13倍アッセイの新しい96ウェルプレートのアリコートで組み合わせる
注:入口あたりの最終的なボリュームは考慮ボリュームの転送中に損失を取るのに十分な量(5.5μl)を準備し、5μlのです。 - 泡を避け、よく混和して、流体を集めるために手短に遠心。
3。チップのロード
注:サンプルとアッセイチップの正確な位置を確保するのが正しいローディングを可能にするために。それは、左上隅にノッチを配置すると便利です。
すべてのアッセイを確認して、サンプルのソリューションは、チップをロードする前に混合される。それは96ウェルプレートでこれを行うとチップをロードする前にプレートをスピンダウンすると便利です。後で参照するためにチップピペッティングマップに注意してください。
未使用のサンプル注入口は2.75μlをサンプルミックス、2.25μlのDNAフリー水で埋めなくてはなりません。
未使用のアッセイの入口は、2.5μlのアッセイローディング試薬および2.5μlの水で埋めなくてはなりません。
気泡を導入しないようにピペッティングしながら最初のピットストップを越えて移動しないでください。
- サンプルとアッセイをロードした後(V入口あたりolume:5μlのは)チップにサンプルとアッセイを読み込むためにLoadミックススクリプト(136x)スクリプトを実行します。
- スクリプトが完了したら、ICFコントローラからチップを取り外します。
- ロードされたチップの裏面から青い保護フィルムをはがします。
- チップ表面のほこりの粒子やゴミを取り除きます。
4。 96.96定量RT - PCR
- BioMarkシステムにチップを転送すると、次の増幅条件を使用します。
55℃2分間、95℃で15秒及び55℃1分間、95℃での40サイクルで10分間、。 - ソフトウェアのデータ収集のための製造業者のプロトコールに従ってください。
5。定量PCR解析ソフトウェアを使用する
RT - PCRの後に独自のソフトウェアでは、増幅曲線、ヒートマップと各ウェルのCt値を提供します。データ解析用ソフトウェアのマニュアルを参照してください。
9。代表的な結果:

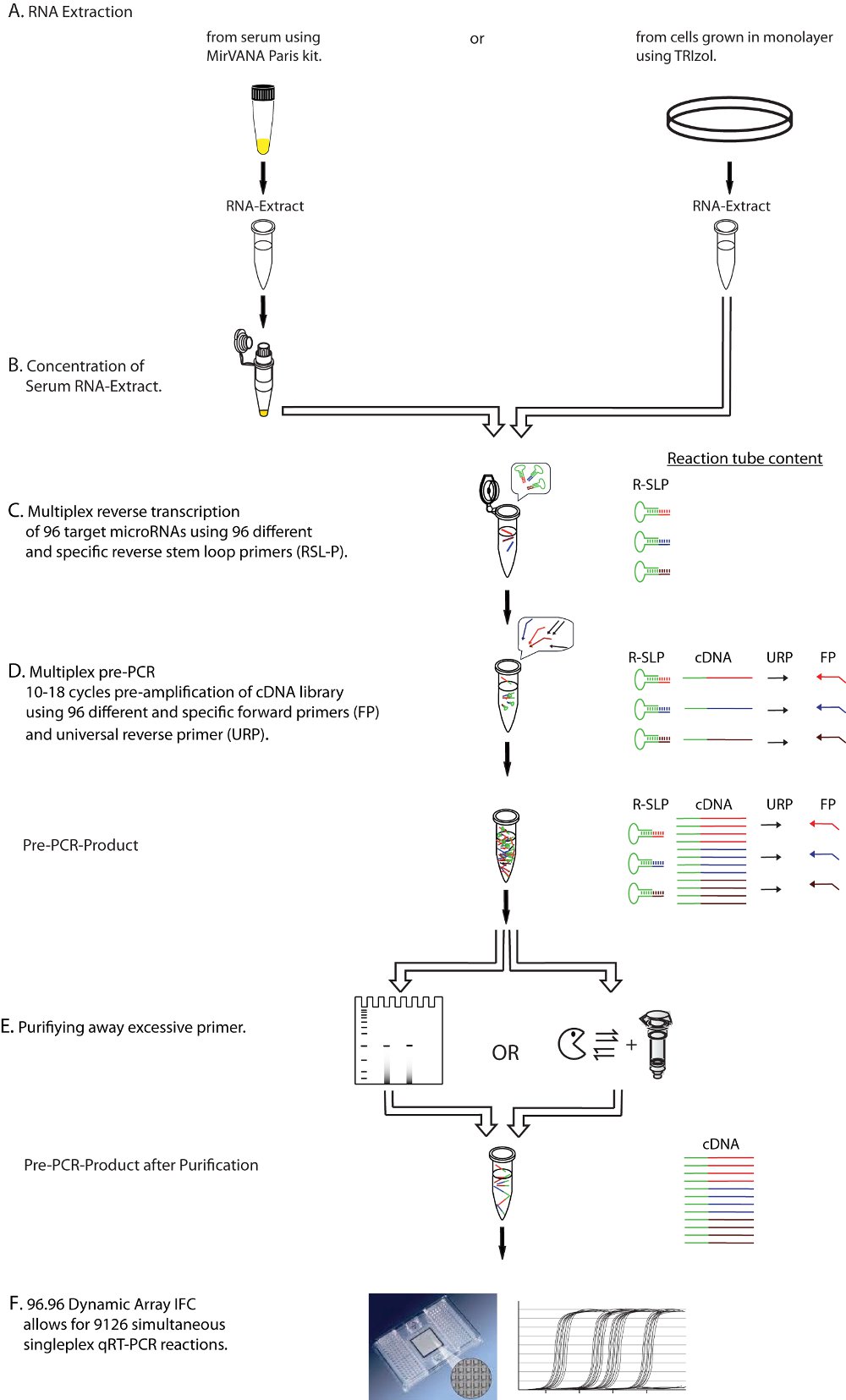
図1。実験的なワークフローの模式図。示されているのは前に多重逆転写(ステップC)と多重プリアンプからの過剰なプライマーの精製(ステップE)(Dステップ)を含むマルチプレックス定量RT - PCRプロトコールのステップバイステップの説明、です。定量RT - PCR用精製したcDNAをテンプレートにその結果をリアルタイムで検出し(ステップF)、。 FP:マイクロRNA特異的フォワードプライマー、URP:ユニバーサルリバースプライマー、R - SLP:マイクロRNAの特定の逆ステムループプライマー。
拡大するにはここをクリックしてください。

図2。あらかじめPCR後の定量RT - PCR鋳型のポリアクリルアミドゲル精製済みのPCR産物のサイズのバンドは排他的にDGCR8欠損WTサンプルではなく、マイクロRNAに見られている- 。96プレックスが後にサンプル(矢印) - / -またはダイサー- / RTとプリアンプの12サイクル。未知の起源(矢頭)と過剰なプライマーの非特異的な製品は、すべてのサンプル(星)で発見されることに注意してください。

図3。多重プリアンプ後の精製が非常に定量RT - PCRの結果の精度を向上させます )Dgcr8へWT MESCの相対的な発現レベルの比較- 。/ - (欠損標準的なマイクロRNA)MESCはより多くのマイクロRNAと偽陽性の2)損失の1)検出を明らかにあらかじめPCR産物のプライマーを離れて精製した後背景- / - Dgcr8の信号。 B)精製した後、DGCR8両方の相対的な発現レベル- / - / WTとダイサー - / - / WTは、偽陽性シグナルの損失を示し、稀Dgcr8独立/ ダイサーに依存する低分子RNA(のmiR - 320、の適切な分類を可能に- 484 -877)5。

図4。フリューハイスループット定量RT - PCR miRNAプロファイリング。サンプル画面は、前立腺癌患者の血清のmiRNAレベルの変化のために画面に96.96フリュー定量RT - PCR mircoRNAプロファイリングの撮影。リアルタイム定量PCR解析ソフトウェアは、増幅曲線、色分けされたヒートマップとサイクル閾値(Ct)を提供しています。
Discussion
miRNAが短い(18〜24ヌクレオチド)、非コーディングの両方不安定メッセンジャーRNA(mRNA)のと阻害の翻訳によって、転写後に遺伝子発現を調節するRNAを、6はヒトの遺伝子の少なくとも3分の1が保存されて含まれているという事実のmiRNAそれらの3'UTRに結合部位および多分化能、増殖およびアポトーシスの遺伝子とmiRNAの明らかな相互作用は、細胞の運命決定、組織の恒常性や癌などの疾患で重要な貢献を示唆している。6,7したがって、正確なmicroRNAの発現プロファイルは広いのです関心。
公開されたマルチプレックス定量RT - PCR miRNA発現プロファイリングのためのプロトコル2をテストするために我々は、ネガティブコントロールとして、小さなRNA欠損MESCを使用していました。 DGCR8 - / - / - -ダイサーしながら細胞は、すべての標準的なマイクロRNAの欠乏している。細胞は正規と非canoncial両方micoRNAsを欠いている8,9
/ - - DGCR8 MES - に野生型のレベルを比較すると、細胞を、我々は、いくつかが10マイクロRNAが野生型細胞11で検出されなかった表現される精度の欠如を発見した。一部ではノックアウト細胞(図3a)に対して相対的な低発現レベルを示した。我々は、精度の不足がsingleplex RT -定量化する前の、2つの連続多重ステップの大規模なプライマーキャリーオーバー(RTとPre - PCR)によって引き起こされるかもしれないという仮説を立てた。しかし、多重プレ増幅は、RNAの初期濃度が限られている場合は特に、信号強度を高めることが必要である。ネイティブポリアクリルアミドゲル上でサイズの選択によりプライマーから事前にPCR産物を離れて精製することにより(図2)、我々はより多くのマイクロRNAとDgcr8両方の偽陽性シグナルの損失を検出することにより、精度の大幅な改善を示すことができる- / -とダイサーを - / -加えて、背景(図3a及び図3b)珍しいDgcr8独立/ ダイサー依存低分子RNA(図3b)11の適切な分類のために許可されて修正された定量RT - PCRのアプローチ。したがって、離れてプレPCR産物からの過剰なプライマーを浄化するために追加のステップは、サンプルあたりの大型マルチプレックスプライマーセットを使用する場合は特に、明らかに有利である。
プレ増幅の最適なサイクル数は、インプットRNA濃度によって決定されます。バランスのないアンダーまたはoveramplifyは、特定の実験の細目によって導かれるべきである。
複数のサンプルを比較する場合は特に1000以上の既知のマイクロRNA、標準を使用すると384ウェルプレートでは、大規模なマイクロRNAプロファイリングのための最適ではない可能性があります。フリュー動的配列IFCは、ナノリットルスケール(6.7 NL)で一回の実験(9216反応)にある97の異なるマイクロRNAに対して96個のサンプルをテストし、ピペッティング操作と必要な化学の大幅な減少と、有効になります。各実行のための解析ソフトウェアには、増幅曲線、色分けされたヒートマップと周期のしきい値(CT)を提供します。同時大規模なプロファイリングは、実験的な分散を低減し、小さなRNAのコントロールを利用する他の正規化戦略を立案、実行平均発現値の正規化、が可能になります。12
事前に割り当てられたTaqManプローブと市販の384ウェルのプラットフォームと比較して、カスタムメイドのプライマーセットを使用してハイスループットプロファイリングプラットフォームの組み合わせは高い実験的な柔軟性を提供しています。
動的配列のプラットフォームとの組み合わせで最適化されたマルチプレックス定量RT - PCRアプローチは成功で同時に384のmiRNA(図4)のレベルの変化で48前立腺癌患者の血清をスクリーニングするとコントロールのスパイク(すなわちsyntheticマイクロRNA)せずにデータを正規化することができました。 11
試料の準備からプロファイリング結果にステップの増加にもかかわらず、説明するアプローチでも限られた出発材料と、時間とmiRNAの発現レベルには大きなサンプルセットをプロファイリングするために費用対効果に優れた高スループット法である。
Disclosures
アランミールはフリュー株式会社の従業員です。そうでなければ、我々は、開示する金融利害関係はありません。
Acknowledgments
我々は、本文についてコメントするためBlellochラボに感謝します。この作品は、からNIH(K08 NS48118とR01 NS057221)からRB、再生医療(CIRM)カリフォルニア工科大学(シードグラントRS1 - 00161、新学部賞RN2 - 00906)とピュー慈善信託するとFMの資金によって支えられてWissenschaftlich UrologischeフラウeVの。
References
- Mitchell, P. S. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 105, 10513-10518 (2008).
- Tang, F. 220-plex microRNA expression profile of a single cell. Nat Protoc. 1, 1154-1159 (2006).
- Gertsenstein, M., Vintersten, K., Behringer, R. Manipulating the Mouse Embryo: A Laboratory Manual. 3, Cold Spring Harbor Laboratory Press. 644-645 (2003).
- Suh, N. MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr Biol. 20, 271-277 (2010).
- Babiarz, J. E., Ruby, J. G., Wang, Y., Bartel, D. P., Blelloch, R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 22, 2773-2785 (2008).
- Babiarz, J. E. Small RNAs: their biogenesis, regulation and function in embryonic stem cells. StemBook. , (2009).
- Friedman, R. C., Farh, K. K., Burge, C. B., Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92-105 (2008).
- Wang, Y., Medvid, R., Melton, C., Jaenisch, R., Blelloch, R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 39, 380-385 (2007).
- Kanellopoulou, C. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 19, 489-501 (2005).
- Thomson, J. M., Parker, J., Perou, C. M., Hammond, S. M. A custom microarray platform for analysis of microRNA gene expression. Nat Methods. 1, 47-53 (2004).
- Moltzahn, F. Microfluidic based multiplex qRT-PCR identifies diagnostic and prognostic microRNA signatures in sera of prostate cancer patients. Cancer Res. 71, 550-560 (2011).
- Mestdagh, P. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 10, R64-R64 (2009).

{kind=link}