

Abstract
Il est devenu de plus en plus évident que la distribution spatiale et le mouvement des composants de la membrane comme les lipides et les protéines sont des facteurs clés dans la régulation de nombreuses fonctions cellulaires. Toutefois, en raison de la dynamique rapide et les petites structures impliquées, une résolution spatio-temporelle très élevée est nécessaire pour rattraper le comportement réel de molécules. Ici, nous présentons le protocole expérimental pour l'étude de la dynamique des protéines et des lipides du plasma membrane marquées par fluorescence dans des cellules vivantes avec une résolution spatio-temporelle élevée. En particulier, cette approche n'a pas besoin de suivre chaque molécule, mais il calcule le comportement de la population en utilisant les molécules d'une région donnée de la membrane. Le point de départ est une imagerie rapide d'une région donnée sur la membrane. Ensuite, une fonction spatio-temporelle complète autocorrélation est calculée en corrélation des images acquises à l'augmentation des retards de temps, par exemple tous les 2, 3, n répétitions. Il est possible de démontrer que la largeurdu pic de la fonction d'autocorrélation spatiale augmente en augmentant le temps de retard en fonction du mouvement des particules par diffusion. Par conséquent, le montage de la série de fonctions d'auto-corrélation permet d'extraire la protéine réelle signifie déplacement quadratique de l'imagerie (DSGI), présenté ici sous la forme de la diffusivité apparente vs déplacement moyen. Cela donne une vision quantitative de la dynamique moyenne des molécules simples avec une précision nanométrique. En utilisant une variante de la GFP étiqueté du récepteur de la transferrine (TfR) et un ATTO488 1-palmitoyl-2-hydroxy-sn -glycéro-3-phosphoéthanolamine (PPE), il est possible d'observer la régulation spatio-temporelle des protéines et des lipides diffusion sur régions membranaires um de taille dans la gamme micro-à-milli-seconde fois.
Introduction
À partir du modèle original "de la mosaïque fluide» par Singer et Nicolson, l'image de la membrane plasmique cellulaire a été continuellement mis à jour au cours des dernières décennies afin d'inclure le nouveau rôle de cytosquelette et de lipides domaines 1,2.
Les premières observations ont été obtenues par récupération de fluorescence après photoblanchiment (FRAP) dévoilement qu'une fraction significative des protéines de la membrane est immobile 5.3. Ces études pionnières, bien que très instructif, ont souffert de la résolution relativement faible dans l'espace (microns) et de temps (en secondes) de configurations du PAF. En outre, étant une mesure de la moyenne d'ensemble, FRAP manque en donnant des informations sur le comportement de la molécule unique.
Dans ce contexte, la possibilité d'étiqueter spécifiquement une molécule unique avec des balises très claires (permettant l'étude du processus de diffusion d'une molécule à la fois) a été très réussie. En particulier, en poussant l'résolution temporelle de l'approche unique de suivi de particules (SPT) à l'échelle de temps de microsecondes, Kusumi, et al. pu accéder aux fonctions inconnues de lipides et de protéines dynamique qui a grandement contribué à la reconnaissance du rôle de base de l'actine-membrane squelette dans la membrane physiologie 6 , 7. Ces résultats ont généré le soi-disant le «piquetage et de clôture» modèle, dans lequel lipides et des protéines diffusion est régie par squelette d'actine. Toutefois, afin d'avoir accès à l'énorme quantité d'informations fournies par SPT nombreuses questions expérimentales doivent être pris en compte. En particulier, la procédure de marquage est typiquement constituée par de nombreuses étapes telles que la production, la purification et l'introduction de l'espèce marquée dans le système. En outre, de grandes étiquettes, comme des boîtes quantiques ou des nanoparticules métalliques, sont souvent nécessaires pour parvenir à l'échelle de temps inférieure à la milliseconde et de la réticulation des molécules cibles par l'étiquette ne peut être évitée dans la plupart des cas. Enfin, de nombreuses trajectoiresdoivent être enregistrés pour s'adapter à des critères statistiques et de façon concomitante une faible densité de l'étiquette est nécessaire pour permettre le suivi.
Par rapport à SPT, la spectroscopie de corrélation de fluorescence (FCS), surmonter un grand nombre de ces inconvénients, représente une approche très prometteuse pour étudier la dynamique moléculaire. En fait, FCS fonctionne bien également avec des étiquettes sombres et denses, ce qui permet d'étudier la dynamique des molécules de protéines étiquetées fluorescentes dans les cellules transfectées de façon transitoire. En outre, il permet d'atteindre des statistiques élevées dans un laps de temps limité. Enfin, malgré la densité «élevé» d'étiquettes FCS fournit des informations sur les molécules simples. Merci à toutes ces propriétés, FCS représente une approche très simple et a été largement appliquées à l'étude de lipides et de protéines dynamique à la fois dans des membranes modèles et dans les cellules vivantes 8-10. De nombreuses approches différentes ont été proposées pour augmenter la capacité de FCS pour révéler les détails de la diffusion moléculaire. Par exemple, il est shpropre que par l'exécution FCS sur les zones d'observation de taille différente, on peut définir un "droit de diffusion FCS" caractéristiques éclairantes cachés de mouvement moléculaire 11,12. En plus d'être varié en taille, le domaine d'intervention a également été dupliqué 13, déplacé dans l'espace le long des lignes 14-20 ou conjugués avec des caméras rapides 21,22. L'utilisation de ces corrélation «spatio-temporelle» les approches, les paramètres biologiques pertinents de plusieurs composants membranaires ont été décrits quantitativement sur les deux membranes modèles et les systèmes biologiques réels, ainsi aperçu rendement dans la membrane organisation spatiale.
Cependant, dans tous les FRAP et applications FCS décrit jusqu'à présent la taille de la zone de convergence représente une limite de la résolution spatiale qui ne peut être surmontée. Plusieurs méthodes d'imagerie de super-résolution ont été développés récemment pour contourner cette limite. Certains sont basés sur la précision de localisation, telles que la microscopie optique de reconstruction stochastique (STORM) <sup> 23,24, microscopie photo-activation de la localisation (PALM) 25, fluorescence PALM (FPALM) 26, et une particule suivi PALM (sptPALM) 27: la quantité relativement importante de photons nécessaires à chaque cliché, cependant, limite la résolution temporelle de ces méthodes à au moins quelques millisecondes, ce qui entrave leur applicabilité in vivo.
En revanche, une alternative prometteuse pour l'imagerie de super résolution ont été ouverts en modulant spatialement l'émission de fluorescence avec des procédés d'épuisement émission stimulés (STED ou transitions réversibles saturables optique de fluorescence (RESOLFT) 28,29). Ces approches se combinent la mise en forme du volume d'observation et au-dessous de la limite de diffraction, avec la possibilité d'utiliser des microscopes à balayage rapide et des systèmes de détection. En combinaison avec l'analyse de fluctuation de fluorescence, la microscopie STED a permis de sonder directement les dynamiques spatio-temporelles à l'échelle nanométrique de lipides et de proteins dans les membranes des cellules vivantes 30,31.
Les mêmes quantités physiques de base de la microscopie STED peuvent être obtenus par une spectroscopie de corrélation de l'image spatio-temporel modifié (TIQUES 32,33) procédé qui est approprié pour l'étude de la dynamique des protéines marquées par fluorescence de membrane et / ou des lipides dans les cellules vivantes et par un microscope commercial. Le protocole expérimental présenté ici se compose de quelques étapes. La première nécessite une imagerie rapide de la région d'intérêt sur la membrane. Ensuite, la pile résultante d'images est utilisée pour calculer les fonctions de corrélation spatio-temporelle moyenne. En adaptant la série de fonctions de corrélation, la «loi de diffusion» moléculaire peut être obtenu directement à partir de l'imagerie sous la forme d'une diffusivité apparente (D app) - vs tracé du déplacement -Moyenne. Cette parcelle est fortement tributaire de l'environnement exploré par les molécules et permet de reconnaître directement les modes de diffusion réelsdu lipide / protéine d'intérêt.
Dans plus de détails, comme indiqué précédemment 34, la fonction d'auto-corrélation spatio-temporelle de la série d'images acquises dépend essentiellement de la dynamique des molécules en mouvement dans la série d'images recueillies (s'il vous plaît noter que le même raisonnement peut être appliqué à l'acquisition d'une ligne où seulement une dimension dans l'espace est pris en compte). En particulier, nous définissons la fonction de corrélation:
 (1)
(1)
où  représente l'intensité de fluorescence mesurée à la position x, y et à l'instant t,
représente l'intensité de fluorescence mesurée à la position x, y et à l'instant t, ![]() et
et ![]() représente la distance dans le plan x etdirections y respectivement,
représente la distance dans le plan x etdirections y respectivement, ![]() représente le décalage dans le temps, et
représente le décalage dans le temps, et ![]() représente la moyenne. Cette fonction peut être exprimée comme suit:
représente la moyenne. Cette fonction peut être exprimée comme suit:
 (2)
(2)
où «N» représente le nombre moyen de molécules dans la zone d'observation, ![]() représente l'opération de convolution dans l'espace, et
représente l'opération de convolution dans l'espace, et  représente l'auto-corrélation de la taille instrumentale. Ce dernier peut être interprété comme une mesure de la façon dont les photons d'un seul émetteur sont répartis dans l'espace en raison de la configuration optique / enregistrement (ce qu'on appelle le Point Spread Function, PSF, gèneRALLYE bien approchée par une fonction gaussienne). Enfin,
représente l'auto-corrélation de la taille instrumentale. Ce dernier peut être interprété comme une mesure de la façon dont les photons d'un seul émetteur sont répartis dans l'espace en raison de la configuration optique / enregistrement (ce qu'on appelle le Point Spread Function, PSF, gèneRALLYE bien approchée par une fonction gaussienne). Enfin,  représente la probabilité de trouver une particule à une distance
représente la probabilité de trouver une particule à une distance ![]() et
et ![]() après un temps de retard
après un temps de retard ![]() . Si l'on considère une dynamique de diffusion, dans laquelle les particules se déplacent au hasard dans toutes les directions et les flux nets ne sont pas présents, cette fonction est également bien approchée par une fonction gaussienne où la variance peut être identifié comme le déplacement quadratique moyen (MSD) de la particule en mouvement . Ainsi, la taille de la fonction de corrélation (également désigné en tant que
. Si l'on considère une dynamique de diffusion, dans laquelle les particules se déplacent au hasard dans toutes les directions et les flux nets ne sont pas présents, cette fonction est également bien approchée par une fonction gaussienne où la variance peut être identifié comme le déplacement quadratique moyen (MSD) de la particule en mouvement . Ainsi, la taille de la fonction de corrélation (également désigné en tant que ![]() ), Peut être définie comme la somme des TMS de particules et la taille instrumental et peut être mesurée par un ajustement gaussiention de la fonction de corrélation pour chaque temps de retard. Le TMS i mesurée peut être utilisée pour calculer une diffusivité apparente des molécules qui se déplacent
), Peut être définie comme la somme des TMS de particules et la taille instrumental et peut être mesurée par un ajustement gaussiention de la fonction de corrélation pour chaque temps de retard. Le TMS i mesurée peut être utilisée pour calculer une diffusivité apparente des molécules qui se déplacent ![]() et un moyen de déplacement
et un moyen de déplacement ![]() en tant que:
en tant que:
 (3)
(3)
 (4)
(4)
Quelques considérations sur le dispositif expérimental utilisé peut guider le lecteur à travers les sections suivantes. Pour exciter sélectivement les fluorophores sur la membrane basale des cellules, nous allons utiliser un éclairage réflexion totale interne (TIR) vivant, en utilisant une fluorescence TIR commerciale (FRBR) microscope (détails peuvent être trouvés dans la section matériel). En outre, afin de recueillir èmee fluorescence, nous allons utiliser un objectif de grossissement (100X NA 1,47, ouverture numérique élevée est nécessaire pour la FRBR éclairage) et une caméra EMCCD (taille physique du pixel sur la puce um 16). Pour atteindre une taille de pixel de 100 nm, nous appliquons une lentille de grossissement supplémentaire de 1.6X. Comme nous le verrons, une résolution temporelle inférieure à 1 ms serait nécessaire pour décrire correctement la dynamique des lipides membranaires rapides en dessous de 100 nm. Afin d'atteindre cette résolution temporelle nous devons sélectionner une région d'intérêt (ROI) plus petit que l'ensemble puce de l'appareil photo (512 x 512). De cette façon, la caméra va lire un nombre réduit de lignes de plus en plus la résolution temporelle. Toutefois, dans ce régime de lecture du temps de trame serait limitée par le temps nécessaire pour transférer les charges de l'exposition à la puce de lecture sur l'appareil et est généralement de l'ordre de 512 millisecondes pour x 512 pixels EMCCD. Pour battre cette limite, une technologie émergente permet le déplacement des ROI lignes seulement au lieu de toute la trame, wvec une réduction effective pratique de la taille de la puce exposée (appelé mode du capteur recadrée dans notre EMCCD). Pour cette configuration, pour être efficace, la puce à l'extérieur de la région d'intérêt doit être couverte par un couple de fentes montées dans le chemin optique. Merci pour cette configuration une résolution de temps d'arrêt à 10 -4 secondes peut être obtenue. S'il vous plaît noter, cependant, que cette approche peut être couplé avec différents montages expérimentaux, comme expliqué dans la section «discussion».
Démonstration de la méthode sera fourni dans les cellules vivantes, en utilisant à la fois un ATTO488 1-palmitoyl-2-hydroxy-sn--glycéro 3-phosphoéthanolamine (ATTO488-EPI) et un variant de la GFP-marqué de récepteur de la transferrine (GFP TfR). Dans le cas de ATTO488 EPI cette approche peut réussir à récupérer une application presque constante de D en fonction de déplacement moyenne indiquant une diffusion pour la plupart gratuites, comme indiqué précédemment 30,35. En revanche, l'ISF-GFP montre une diminution D
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Système d'étalonnage de 1
- Point Spread Fonction étalonnage (PSF)
- Diluer 10 ul de solution de 30 nm de billes fluorescentes (environ 5 pM) dans 90 ul d'eau distillée, puis la solution sonication pendant 20 min. Couper un morceau de gel d'agarose (3%) et le dépôt de 10 ul de la solution sur le dessus du gel carré (1 cm x 1 cm). Renverser le morceau de gel sur la vitre fond d'un plat de 2 cm de Pétri et presser la baisse sur la vitre.
- Tournez sur la configuration d'acquisition, mettre l'échantillon dans le support, définition de l'exposition de l'appareil photo et EMgain (100 ms et 1000 sont de bons paramètres, mais d'optimiser en fonction du système) et attendent l'appareil refroidir.
- Réglez l'exposition de la caméra à 100 ms, caméra EMgain à 1000, le mode d'acquisition à l'image de transfert, 100 répétitions et l'enregistrement automatique de réglage.
- Utilisation de l'oculaire et l'orientation de la lumière transmise à la frontière du gel et ensuite passer l'objectif au centre du gel, la mise au point et start la procédure d'alignement laser (à LAS AF, sélectionnez "configuration de la FRBR» et suivez la procédure d'alignement automatique).
- Trouver un champ de vision avec des taches simples isolées, précisément l'accent sur la place lumineuse (qui représente généralement perles total) comme référence, l'acquisition de 100 images et répétez l'étape 5-6 fois dans le but d'acquérir plusieurs taches simples.
- Importez la série acquise à un programme de traitement de données et la moyenne de la pile dans le temps (figure 1A) et sélectionnez une seule bille isolé. Prenez soin de choisir les plus petits pour éviter des agrégats de particules.
- Monter la distribution sélectionnée d'intensité (un exemple de profil unique de perles est présenté dans la figure 1B) avec une fonction gaussienne en utilisant la commande "gaussfit" (dans les outils ICS-Matlab dans les matériaux en Matlab). Vérifiez la qualité de l'ajustement en inspectant les résidus obtenus (un exemple de profil gaussien équipée avec les résidus correspondants est présenté in Figure 1B).
- l'étalonnage de l'appareil photo
- Allumez l'appareil photo et attendez que l'appareil refroidisse. Réglez cadre de l'acquisition de la caméra, (c'est à dire, pour l'appareil utilisé, nous mettons l'exposition à 0,5 ms, caméra EMgain à 1000, le mode d'acquisition en mode courte, la taille de la ROI de 32 x 128, 10 000 répétitions) et lancer l'acquisition de l'arrière-plan de la caméra signaux.
- Importation acquis série de trames à un programme de traitement de données. Calculer et contrôler l'intensité moyenne de chaque pixel afin de vérifier que le fond de l'appareil est sensiblement plane dans la région sélectionnée de la puce. En mode courte, enlever la première et les dernières lignes horizontales (3 à 10 en fonction de la taille de la ROI) pour chaque trame, car le fond de l'appareil est généralement sollicité dans les lignes de bordure.
- Créer un histogramme des valeurs (également défini de niveau numérique, DL) dans les images acquises pile (en utilisant la commande 'hist' dans Matlab) et tracer le logarithmede la fréquence résultant (en utilisant la commande semilogy dans Matlab). Un exemple de la distribution de DL caméra arrière-plan est présenté à la figure 2.
REMARQUE: Si l'appareil fonctionne bien, l'intrigue se montrer un pic approximativement gaussienne (un profil parabolique en échelle logarithmique) représentant la distribution des valeurs associées à photon zéro suivie d'une décroissance exponentielle (une droite de pente négative en échelle logarithmique ) qui représente la distribution des valeurs associées à une photon (figure 2). En particulier, le centre et la variance de la fonction gaussienne représente le décalage de la caméra et de l'erreur, respectivement, tandis que la constante de décroissance exponentielle de la pièce représente une estimation de la DL attribuée par l'appareil à chaque photon unique. Dans Matlab utiliser la section "CalibrateCamera" de l'écriture dans les matériaux de support. - Répétez l'opération pour tous les appareil photo sélectionnée EMGain et Gain.
2. étiquetésPréparation cellulaire
- Pour préparer les liposomes de lipides nécessaires pour l'incorporation 36, dissoudre séparément une mg de DOPE (1,2-sn dioleoyl- -glycéro-3-phosphoéthanolamine), 1 mg de DOTAP (1,2-dioléoyl-3-triméthylammonium-propane), et 1 mg de PPE-ATTO488 dans 1 ml de chloroforme. Mélanger 0,5 ml de solution à filer, de 0,5 ml de solution DOTAP, et 25 ul de solution PPE-ATTO488 et sèche sous vide pendant 24 h. Ajouter 0,5 ml de tampon HEPES 20 mM, vortex pendant 15 min et soniquer pendant 15 min à 40 ° C.
- Pour préparer la cellule, laver 3 fois avec du PBS un plat de p100 de confluence CHO-K1 (ovaire de hamster chinois), ajouter 1 ml de trypsine et de stocker dans un incubateur pendant 5 min. Suspension de cellules individuelles d'addition de 9 ml de milieu DMEM / F12 supplémenté avec 10% de FBS et ensemencer 150 ul de solution de cellules dans une boîte de Pétri contenant 800 ul du même milieu.
- Conserver dans un incubateur pendant 24 heures à 37 ° C et 5% de CO 2. Pour l'incorporation des lipides, remplacez milieu cellulaire avec 500ul de milieu sans sérum; après 30 minutes, ajouter 2 ul de solution de liposomes; après 15 minutes de lavage avec PSB et ajouter un nouveau milieu DMEM / F12 pour l'imagerie.
- Pour la transfection, les cellules transfecter selon le protocole Lipofectamine (instructions du fabricant) à l'aide TfR-GFP plasmide et magasin de 24 heures dans l'incubateur avant l'imagerie.
3 Acquisition de données
- préparation de l'installation
- Afin de thermostat microscope, 24 heures avant l'expérience allumer l'incubateur.
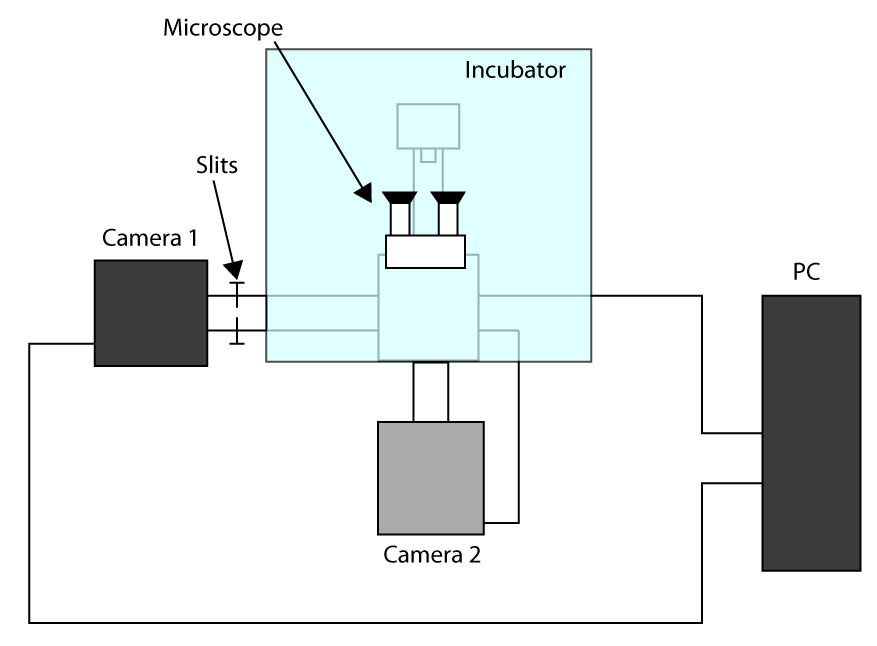
- Pour appliquer l'heure d'acquisition le plus rapide possible, le travail en mode capteur recadrée (voir introduction) et utiliser un premier appareil d'imagerie (appareil photo 1) et deuxième caméra pour sélectionner la cellule (l'appareil photo 2). Un schéma de la configuration d'installation est présentée dans la figure complémentaire S1. Ensuite, pour aligner le tour de deux caméras sur le microscope et attendre que les caméras se refroidir.
- Situé sur les deux appareils les paramètres pour l'imagerie lumière transmise (c.-à-
- Mettre les échantillons dans le support et mise au point avec l'oculaire, envoyer la lumière à la caméra 1 et poussez doucement les fentes permettant la lumière que sur le retour sur investissement utilisés pour l'imagerie cellulaire (ici un retour sur investissement de 32 x 32 pixels).
- Déplacer une cellule dans la région sélectionnée et envoyer la lumière à la caméra 2, puis dessinez un retour sur investissement dans le logiciel Camera Control 2 afin d'avoir une référence.
- Imaging (figure 3A)
- Tout d'abord, aligner le laser de la FRBR selon la procédure de votre configuration. Dans notre configuration, sélectionnez le 'setup FRBR »et lancez la procédure d'alignement automatique. Lorsque le laser est aligné mis 70 nm de profondeur de pénétration (environ 70 °).
- Réglez le temps d'exposition à 70 ms et EMGain à 100 à la fois sur les caméras 1 et 2; Ensuite, sélectionnez une cellule en utilisant la caméra 1, puis envoyer la lumière sur la caméra 2 et mise au point précise de la membrane cellulaire. Réglez l'exposition minimale sur camera 2, 1000 EMGain, mode du capteur courte, 10 5 répétitions et définir l'enregistrement automatique comme les fichiers FITS (Flexible Image Transport System, un format qui peut être facilement géré).
- Lancer l'acquisition d'enregistrer la série d'images. Relâchez le gain et le mode recadrée pour permettre la stabilisation de la température avant d'acquérir une nouvelle cellule, puis répétez les deux dernières étapes pour acquérir 8-10 cellules.
4 Calcul du déplacement quadratique moyen de l'imagerie (i MSD)
NOTE: Le protocole suivant peut être appliqué directement aux données brutes. Dans le même temps, l'ensemble du protocole est valide pour l'acquisition de données simulées à la fois dans Matlab et dans SimFCS. Le lien vers les tutoriels correspondant peut être trouvé dans la section "Matériaux de.
- Calcul par Matlab
- Importez la série acquise à Matlab en utilisant un script de ImportImageSeries. Calculer l'intensité moyenne de chaque image dans le temps en utilisant la command moyenne sur les deux premières dimensions et utiliser parcelle pour voir le vecteur résultant.
- Si plus de 10% de photoblanchiment est présent, jeter la série ou supprimer la première partie d'entre eux. Si elle est inférieure, essayer de corriger l'effet sur la fonction de corrélation en soustrayant à chaque image son intensité moyenne, comme le montre avant 37.
- Calculer l'intensité moyenne de chaque pixel en utilisant le moyen pour la troisième dimension et de voir l'image résultante.
Remarque: Une attention particulière est nécessaire afin d'éviter des corrélations artefacts. En fait, comme le montre déjà des techniques semblables 38, les bordures de cellules ainsi que sur des vésicules de discussion pourrait introduire une forte corrélation. Si l'inspection de l'image moyenne révèle les bordures de cellules ou de vésicules de discussion, essayer d'exclure la région participait pas jeter l'acquisition. Pour corriger l'effet de cette structures immobiles soustraire l'intensité temporelle moyenne de chaque pixel 39. - Calculer til corrélation spatio-temporelle (G (ξ, χ, τ)) en utilisant la fonction CalculateSTICScorrfunc. Retirer G (ξ, χ, 0), car la corrélation à cause du bruit de coup de feu en régime de faible lumière domine G (0,0,0); la corrélation en raison de la détection domine le G (± 1,0,0), et le mouvement des particules au cours de la durée d'exposition peut se déformer G (ξ, χ, τ) pour τ = 0 en augmentant la taille mesurée (cet effet disparaît pour τ > 0) 34.
- Moyenne G (ξ, χ, τ> 0) en utilisant un temps-bin logarithmique pour réduire le bruit en utilisant la fonction "LogBinStack" dans le soutien matériel et puis poser le G résultant (ξ, χ, τ) en utilisant la fonction "gaussfit" de les outils ICS-Matlab dans les matériaux à récupérer le i MSD (la deuxième colonne du tableau qui en résulte).
- Tracer la σ obtenu de la taille (de τ) 2 (i MSD) en fonction du temps. Si les données sont trop bruyants, essayez d'augmenter le nombre de acquLes cadres IRED, augmenter la puissance du laser, en moyenne, plus G (ξ, χ, τ) en même temps.
- Calcul par SimFCS
- Ouvrez les fichiers acquis avec ImageJ utilisant BIOFORMAT importateur plugin et sauver série acquis que la séquence Tiff.
- Ouvrir outil SimFCS et sélectionnez la RICS et sélectionnez Fichier> Importer des images multiples (supplémentaire Figure S2).
- Sélectionnez Fit, insérer les paramètres d'acquisition correctes et fermer la fenêtre en forme (supplémentaire Figure S3).
- Sélectionnez Affichage> Intensité Moyenne> CH1 et vérifier la présence de photoblanchiment (complémentaire figure S4).
- Si plus de 10% de photoblanchiment est présent jeter la série ou si il est possible charge à nouveau la séquence d'images de retirer la première partie de la série.
- Si le blanchiment est inférieur à 10% sélectionnez Outils> i MSD> Paramétrer, vérifie 'utilisation moyenne mobile », situé dans le panneau de ROI sur la gauche uneombre de cadre à l'attention payer moyenne mobile que le temps correspondant est plus élevé que le temps caractéristique de diffusion (pour particules se déplaçant à 1 pm 2 sec -1 un temps de 10 sec est une bonne moyenne mobile)
- Sélectionnez Outils> DSGI> Calculer i MSD (figure complémentaire S5) et en forme et exporter le i MSD de la bloc-notes (Figure supplémentaire S6).
5 Calcul de la loi Diffusion de la i MSD
- Montez les quelques premiers points d'extrapoler l'interception (σ 0 2) (5 points sont généralement suffisant, mais plusieurs points peuvent être installés s'ils présentent un comportement linéaire) et de comparer cette valeur avec le PSF mesurée précédemment 2. Si elles sont comparables, la dynamique des fluorophores isolés sont respectées. En revanche, si σ 0 2 2 >> PSF tenter d'acquérir plus rapidement pour s'assurer quedynamique pas cachés sont présents 34.
- Calculer la diffusivité apparente (app D) et le déplacement moyen (R) en utilisant les équations 3 et 4 (voir Introduction).
- Terrain application en fonction de R pour obtenir un droit de diffusion comparable à ce qui est mesuré avec une tache variation en fonction FCS 12 (figure 3D) D.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Afin de calibrer la taille instrumental, l'image d'un seul nano-billes fluorescentes peut être mesurée comme décrit dans le protocole de l'étape 1.1. Une image de fluorescence typique de ces billes est présenté sur la figure 1. L'installation de distribution d'intensité d'une fonction gaussienne 2D redonne bonne résidus et permet de mesurer la taille instrumental à 270 nm. Cette valeur est en bon accord avec la limite de diffraction attendue estimée par l'équation de Rayleigh. Cet étalonnage n'est pas nécessaire pour la mesure de la dynamique des particules, mais il est nécessaire de mesurer la taille des particules apparent.
Une distribution typique de la fréquence de la caméra arrière-plan est présenté à la figure 2. Le pic à environ 180 DL est due à la réponse de la caméra à aucun photon, et elle représente la contribution de numérique analogique (AD) convertisseur. Cette contribution peut être approchée comme une distribution gaussienne pour estimer le décalage et la varianceintroduit par l'enregistrement du signal. Au-dessus de 200 DL la distribution numérique de niveau devient exponentielle (linéaire en échelle logarithmique) et représente la réponse moyenne de la caméra à un seul photon. Le montage de ce cadre avec une distribution exponentielle permet les mesures de la DL moyenne attribuée à chaque photon unique. Le plus élevé est le rapport entre la DL moyenne attribuée à chaque photon et l'erreur de convertisseur AD, plus faible sera le bruit dans la fonction de corrélation calculée. En outre, la réponse moyenne de photon unique permet l'estimation de la gamme dynamique de la caméra.
Un schéma de la procédure expérimentale complète est résumée sur la figure 3 et une vue de l'insertion dans l'EPI-Atto488 la membrane est représentée sur la figure 4A. Une image TIRF représentant de la membrane basale des cellules CHO a marquées avec Atto488-EPI est présentée sur la figure 4B. Plusieurs points lumineux peuvent être présents en dehors de la cellule en raison de liposomes empilés sur le verre. Ils peuvent être éliminés par la sélection d'un retour sur investissement sur une portion de membrane essentiellement uniforme de la fluorescence (par exemple., La membrane plasmique cellulaire). Comme prévu, le droit de diffusion mesurée (figure 4C) pour ce lipide est plat, ce qui indique une diffusion pour la plupart gratuites, comme indiqué précédemment par des mesures STED-FCS 30,35. Il est à noter que toutes les valeurs de déplacement indiqués sont en dessous de la limite de diffraction, indiquant clairement la capacité de cette approche à super-résolution déplacements moléculaires moyens bien en deçà de la limite de diffraction et jusqu'à quelques dizaines de nanomètres.
Une schématisation de TfR-GFP insertion de dimère dans la membrane est représentée sur la figure 5A. De nombreuses études ont montré que la queue cytoplasmique de ce récepteur interagit avec le squelette de la membrane, ce qui à son tour agit comme une barrière pour la mobilité de 12,40 récepteur. Une image de la FRBR représentant d'une cellule CHO exprimant TfR-GFP est presented sur la figure 5B. Des cellules de faible intensité de fluorescence doivent être préférés, comme la membrane est plus proche de l'état natif et la probabilité d'artefacts liés à la sur-expression est réduite au minimum. En outre, la partie centrale de la cellule doit être évitée, car les effets de fluorescence hors foyer (à partir de cytoplasme, par exemple) peuvent être présents. Comme prévu, le droit de diffusion mesurée (Figure 5C) pour TfR-GFP montre un premier comportement plat en dessous de 100 nm, avec une application moyenne de D d'environ 0,7 um 2 sec -1, suivi par une diminution rapide conséquente diffusivité apparente jusqu'à 0,2 um 2 sec -1 (la valeur généralement mesurée par diffraction limitée FCS 12). Ce résultat montre que notre approche peut facilement mesurer le déplacement moyen de protéines GFP marquées avec une résolution de quelques dizaines de nanomètres. De plus l'échelle spatiale à laquelle l'application de D commence à diminuer ensembles la caractéristiqueéchelle spatiale de la protéine confinement partiel par le squelette de la membrane autour de 120 nm, en accord avec les estimations antérieures 6.

Figure 1: Calibration de Point Spread Function. (A) de l'image Pseudocouleur de bourrelet et perles isolées agrégats. (B) terrain 3D du profil d'intensité d'un bourrelet isolé montre un profil gaussien bien défini. (C) Fit de la distribution d'intensité par une fonction gaussienne (panneau supérieur) avec les résidus correspondant (panneau inférieur). Le bon accord entre la distribution ajustée et le profil d'intensité mesurée est aussi une preuve que la PSF instrumentale peut être approchée par une fonction gaussienne. S'il vous plaît cliquer ici pour vi ew une version plus grande de cette figure.

Figure 2: étalonnage de la réponse de l'appareil à photons uniques. Cette figure montre la distribution numérique de niveau (DL) pour appareil photo arrière-plan dans un ROI de 32 x 128, une exposition de 0,5 ms, en mode capteur recadrée. Le pic à environ 180 DL représente la réponse de la caméra à aucun photons. En particulier, il représente la contribution du convertisseur analogique numérique (AD) et peut être approchée par une fonction gaussienne pour estimer le décalage et la variation introduite par l'enregistrement du signal. Au-dessus de 200 DL de la distribution des niveaux numériques devient exponentielle et représente la réponse moyenne de la caméra à un seul photon. La mesure de ces paramètres permet d'estimer la densité de photons qui sont enregistrés lors de l'acquisition.s / ftp_upload / 51994 / 51994fig2highres.jpg "target =" _blank "> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 3 La schématisation de la méthode. (A) l'échelle de formation d'image du champ de la caméra EMCCD est appliquée pour atteindre une résolution inférieure à la milliseconde, tout en TIRF microcopie est exploitée pour assurer le sectionnement optique précise de la membrane plasmique. (B) La pile résultante d'images est autocorrélation pour calculer la moyenne spatiale fonction de corrélation -temporal. Cette fonction de corrélation est bien approximée par une fonction de Gauss (voir l'introduction) et il s'étend dans le temps en fonction des déplacements de particules (C). Ainsi, dans le but de quantifier l'étalement de la fonction de corrélation en raison du déplacement moléculaire, le montage avec un Gauss ian fonction est exécutée. Cela permet la mesure de la «loi de diffusion» moléculaire directement à partir de l'imagerie, sous la forme de la diffusivité apparente vs tracé du déplacement moyenne. (D) Merci pour cette parcelle, les modes de diffusion moléculaire peuvent être directement identifiés sans avoir besoin d'un modèle interprétatif ou des hypothèses sur la l'organisation spatiale de la membrane. En effet, les molécules diffusant librement affiche une diffusivité apparente constante que leur mobilité ne dépend pas de l'échelle spatiale de la mesure. En revanche, les molécules partiellement confinés affiche une diffusivité apparente assez constante pour des déplacements plus petits que la taille de confinement, une diffusivité baisse pour les échelles spatiales plus grandes que la taille de confinement. Ainsi, l'apparition d'une réduction de la diffusivité apparente peut être interprétée comme une empreinte digitale de confinement transitoire, tandis que l'échelle spatiale liée peut être utilisée pour estimer l'extension spatiale de l'accouchement. .jove.com / fichiers / ftp_upload / 51994 / 51994fig3highres.jpg "target =" _blank "> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 4 ATTO488 EPI droit de diffusion dans les membranes des cellules vivantes (A) Représentation schématique de l'insertion ATTO488 EPI dans la membrane de la cellule (B) l'image de la FRBR de CHO membrane basale marqué avec ATTO488-EPI:.. Un (boîte rouge) de retour sur investissement est sélectionné dans une partie essentiellement uniforme de la cellule, évitant bordure de la cellule et des taches très fluorescentes. (C) La loi de diffusion mesuré dans la ROI sélectionnée montre un comportement plat confirmant un modèle de diffusion gratuite pour cette composante. S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.

Figure 5 TfR-GFP loi de diffusion dans les membranes des cellules vivantes (A) Représentation schématique de l'insertion TfR-GFP dans la membrane cellulaire. La queue cytoplasmique du récepteur interagit avec le squelette de la membrane, qui agit comme une barrière à la mobilité de récepteur (B. ) image de la FRBR de CHO exprimant TfR-GFP: un retour sur investissement est sélectionné préférant cellules exprimant de faibles afin d'éviter des artefacts dus à la surexpression (C) La loi de diffusion de l'ISF (points noirs), contrairement PPE (ligne grise, prise de la figure 4),. montre le comportement typique de diffusion partiellement confiné où une première partie plate est suivie d'une diminution en application de D. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Suivi de particules unique (SPT) représente l'une des stratégies les plus courantes pour étudier la dynamique moléculaire et il a le grand avantage de mesurer les trajectoires des particules. Cela permet à son tour le comportement de palpage de même quelques particules marquées dans un système complexe. Toutefois, pour atteindre cet avantage SPT a généralement besoin d'une faible densité de la sonde et des étiquettes très brillantes. En particulier, pour obtenir haute résolution temporelle (plage de microsecondes) sonde inorganique sont généralement nécessaires (par exemple, les points quantiques ou nanoparticules métalliques): dans ce cas, une procédure complexe de la production, l'étiquetage et l'insertion dans le système est nécessaire. Par rapport à la méthode actuelle SPT présente quelques avantages clés. Tout d'abord, cette approche peut être utilisée en conjugaison avec les protéines fluorescentes. Ainsi, par rapport à SPT, une résolution temporelle plus élevée est atteinte (sur le même label), grâce à la faible quantité de photons nécessaires 34. Plus en détail, cette propriété permet de pousser la résol temporelleution dessous 10-3 sec également l'utilisation de protéines fluorescentes pouvant être codées, et cette échelle de temps donne un accès exclusif à la dynamique d'échelle nanométrique de constituants membranaires. Enfin, il est intéressant de noter que les lois de diffusion moléculaire sont décrits par l'analyse de la fonction complète de corrélation espace-temps, sans avoir besoin de suivre chaque molécule.
La comparaison avec FCS base-STED est également intéressant. Dans une mesure STED-FCS le temps de transit moyen des molécules permettant de diminuer les volumes d'observation est mesurée par la corrélation temporelle du signal de fluorescence. Ceci permet l'obtention d'une mesure locale de la dynamique moléculaire aussi sous la limite de diffraction. Dans l'approche présentée le droit de diffusion est évaluée comme la moyenne de toutes les particules se déplaçant dans la ROI sélectionnée, observée au moyen de la norme, limitée par la diffraction, le volume d'observation. Cependant, les résultats démontrent rapporté que cette méthode n'est pas limitée par la diffraction, mais seulement par ee résolution temporelle disponible. En effet, si une acquisition limitée par la diffraction est utilisé pour détecter les fluctuations (de manière analogue à ce qui se fait dans d'autres techniques de super-résolution, comme PALM et STORM), les déplacements moléculaires bien en dessous de la limite de diffraction peut être (directement) calculé, comme déjà démontré à l'aide de STICS pour mesurer les flux moléculaire 32. De plus, contrairement STED-FCS, cette approche peut être facilement appliquée à un large éventail de configurations de microscopie commerciales et existants, tels que les microscopes à balayage de trame ou microscopes à base de caméra à champ large. Il est digne de mention que les mesures STED-FCS des lois de diffusion moléculaire doit impérativement un étalonnage fluorophore dépend de la taille de la taille instrumentale. À l'opposé, la mesure présentée ici ne nécessite pas un étalonnage du système (uniquement nécessaire pour l'estimation de la taille des particules).
La résolution réelle à la mesure des déplacements de particules par le procédé présenté depend de la précision avec laquelle nous pouvons mesurer la fonction de corrélation. Par conséquent, il n'est pas intrinsèquement limitée par la diffraction, de manière analogue au cas où le SPT résolution dépend de la précision du "image" de la particule est mesuré. Pour mesurer une corrélation significative en moins de 1 min pour les expériences proposées, habituellement peu de photons (photons) au-dessous de 10 pour chaque particule dans chaque trame sont suffisants. En effet, la contribution de l'ensemble des particules observées en même temps quand on fait la moyenne de la fonction de corrélation est calculée, même si les particules ne sont pas isolés. Cette propriété est intrinsèque des méthodes de corrélation de fluctuation et permet d'utiliser des étiquettes sombres et denses, telles que les protéines fluorescentes transfectées dans des cellules vivantes.
Dans cette optique, il apparaît clairement que le déplacement minimal mesurable dépend de la diffusivité de la particule et à la résolution temporelle de la configuration de l'image. A titre d'exemple, s'il vous plaît envisager la diffusion des molécules de la membrane cellulaire,où la diffusivité maximale mesurée pour des protéines ou des lipides est d'environ 5 um 2 s -1. Dans ces conditions, nous avons besoin d'une résolution temporelle de l'ordre de 10 -4 sec pour attraper un déplacement moyen de 50 nm. Cette résolution temporelle peut être obtenue par les microscopes à balayage rapide le long des lignes simples ou par caméra EMCCD rapide, où la résolution de temps coïncide à la durée d'exposition, comme l'a montré ici.
Une exigence supplémentaire essentielle pour cette méthode de décrire avec précision la dynamique moléculaire est un échantillonnage spatial correct. En effet, afin d'adapter la fonction de corrélation nous avons besoin d'un échantillonnage spatial (taille du pixel) inférieure à la taille de la PSF instrumentale. Dans la plupart des microscopes commerciaux (champ confocale ou étendu), la taille de la PSF s'étend de 200 nm à 500 nm (principalement en fonction de l'ouverture numérique de l'objectif sélectionné et de la longueur d'onde utilisée) et qui peut être facilement mesuré par un essai d'étalonnage en utilisant nano billes fluorescentes de taille. Thus, une taille de pixel de 70-150 nm (3 fois plus faible que la taille instrumental) peut suffire. Toutefois, la taille des pixels peut être adapté pour le système en cours d'étude en tenant compte d'une règle simple: diminuer la taille des pixels, plus la précision sur la description de la fonction de corrélation. En outre, la taille minimale de l'image à acquérir doit être au moins 3 fois plus grand que le déplacement maximal de l'intérêt (plus la taille instrumental). Ceci est nécessaire afin d'atteindre une bonne convergence de l'algorithme d'ajustement et un échantillon statistiquement significatif de déplacements moléculaires. A titre d'exemple, pour étudier les déplacements moléculaire moyenne inférieure à quelques centaines de nanomètres (par exemple, 200 nm) d'une taille d'image de quelques microns est suffisante. En outre, le nombre total de pixels (en tenant constantes la taille de pixel) impacts sur la qualité de la fonction de corrélation. En fait, une image plus grande permet en moyenne plus d'informations dans la fonction de corrélation, même si au détriment du temps résolution. En ce qui concerne le système basé sur une caméra utilisée ici, s'il vous plaît noter que la taille physique du pixel sur la puce est fixée. Par conséquent, la diminution de la taille des pixels du signal diminue en chaque pixel (qui dépend du carré de la taille de pixel), diminue le champ de vision, et nécessite une puissance de grossissement. D'autre part, dans un système de balayage, où la zone d'observation est fixe, ce qui réduit la taille des pixels se traduit généralement par une augmentation du nombre de pixels, au détriment de la résolution temporelle.
Peu de détails sur le détecteur utilisé doivent être discutés. A la différence des détecteurs de photons uniques, les systèmes de EMCCD mesurer une intensité moyenne (niveau numérique, DL) qui n'est pas directement proportionnelle à la lumière collectée par la présence d'un décalage. Même si ce décalage est faible par rapport à la gamme dynamique de la caméra (quelques centaines par rapport à deux bits 16 à 16 de lecture) et négligeable dans de nombreuses expériences où les photons sont collectées, il doit être pris en compte pourobtenir une normalisation correcte de la fonction de corrélation. En outre, le décalage peut être utilisé comme une référence dans des conditions de faible luminosité afin d'identifier la quantité de signal recueilli. En outre, afin d'estimer le montant moyen de photons qui sont collectées lors de l'acquisition, le niveau numérique moyenne associée à chaque photon collecté doit être mesurée. Cette quantité peut être récupéré par d'exposer l'appareil à une intensité lumineuse très faible (par exemple, la lumière de fond dans la salle); en fait, dans ce cas, on peut raisonnablement supposer que les photons atteignent juste simples de la caméra, c'est à dire l'intensité mesurée peut être reliée à zéro ou un photon seulement.
Enfin, nous commentons sur la façon dont certains systèmes d'acquisition de remplacement (c'est à dire, différentes configurations de microscopie) peuvent être utilisés pour effectuer les mesures présentées. Tout d'abord, la "W'factor dans l'équation 2 (qui représente l'autocorrélation de la PSF instrumentale) peut être adapté à til système d'acquisition particulier utilisé afin d'adapter la fonction de corrélation expérimentale. Comme montré précédemment 34, un cas simple est l'acquisition whit un microscope à balayage laser lorsque la vitesse de balayage est sensiblement plus élevé que la dynamique des particules. Dans un tel cas, en effet, le mouvement des particules au cours du temps d'acquisition (c'est à dire, la durée de ligne) peut être considéré comme négligeable, et la fonction de corrélation est bien approximée par une fonction gaussienne. Dans le cadre des nouvelles technologies d'imagerie, une approche intéressante est basé sur la possibilité de produire des très minces feuilles de lumière (1-2 um) à travers l'échantillon 41. La nappe de lumière permet l'illumination sélective d'un plan unique (Single plan illumination microscopie, SPIM) dans l'échantillon et, associé à un système d'acquisition par caméra, rapide sectionnement optique en 3D 42. En raison de ces caractéristiques, SPIM a été conjugué avec succès FCS 43 et pourrait représenter une valiOutil de d pour prolonger l'analyse présentée à des environnements 3D, tels que le cytoplasme ou le noyau des cellules vivantes.
En résumé, à partir d'un point de vue expérimental, cette approche exige que l'accès à un microscope équipé d'un module d'acquisition rapide. La protéine d'intérêt peut être marquée avec une protéine fluorescente ou fluorophore organique, ce qui permet aussi à l'imagerie multicolore. Dans ce contexte, nous envisageons la possibilité d'utiliser l'analyse croisée i MSD pour sélectionner des sous-populations de molécules et de révéler les interactions et co-diffusion sur les membranes de cellules vivantes. Enfin, nous pensons que cette approche peut représenter un outil puissant pour étudier les protéines et / ou des lipides subissent le partitionnement dynamique dans nanodomaines sur la membrane plasmique. Dans ce cas, la taille très variable et durée de vie des nanodomaines introduire un niveau supplémentaire de complexité en données réelles qui exigerait de nouvelles implémentations méthodologiques, y compris l'imagerie en 2 couleurs, localel'analyse (par exemple, paire corrélation 2D) et / ou l'anisotropie de fluorescence.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| iXon Ultra 897 | Andor | DU-897U-CS0 | |
| Solis | Andor | ||
| CHO-K1 | ATCC | CCL-61 | |
| ATTO 488 labeled PPE | ATTO-TEC GmbH | AD 488-151 | |
| DOPE | Avanti Polar Lipids, Inc. | 850725 | |
| DOTAP | Avanti Polar Lipids, Inc. | 890890 | |
| 100x Penicillin-Streptomycin-Glutamine | Gibco | 10378-016 | |
| DMEM/F-12 | Gibco | 21331 | |
| FBS | Gibco | 10082147 | |
| HEPES | Gibco | 15630-106 | |
| PBS | Gibco | 10010-023 | |
| SimFCS 3.0 | Globals Software | the software can be downloaded here: http://www.lfd.uci.edu/globals/ | |
| DMI6000 with TIRF modulus | Leica | ||
| LAS AF | Leica | ||
| Lipofectamine 2000 | Lipofectamine | 11668019 | |
| Matlab | MathWork | ||
| ImageJ | NIH |
| Name | Company | Catalog Number | Comments |
| C-terminal GFP tagged Tranferrin Receptor | OriGene | RG200980 | |
| Agar | Sigma Aldrich | A5306 | |
| Chloroform | Sigma Aldrich | 528730 | |
| Latex beads, fluorescent yellow-green, 30 nm | Sigma Aldrich | L5155 | |
| SONICA Ultrasonic Cleaners | SOLTEC | ETH S3 | |
| Petri Dishes | Willco | GWSt-3522 | |
| Bio-Format importer for Matlab | http://www.openmicroscopy.org/site/support/bio-formats5/users/matlab/ | ||
| ICS-MatLab Tools | https://www.cellmigration.org/resource/imaging/software/ICSMATLAB_28-02-06.zip | ||
| Simulation by Matlab Tutorial | https://www.cellmigration.org/resource/imaging/icsmatlab/ICSTutorial.html | ||
| Simulation by SimFCS Tutorial | https://www.cellmigration.org/resource/imaging/ppt-pdf/RICS%20Simulations.ppt |
References
- Engelman, D. M. Membranes are more mosaic than fluid. Nature. 438 (7068), 578-580 (2005).
- Vereb, G., et al. yet structured: The cell membrane three decades after the Singer-Nicolson model. Proc. Natl. Acad. Sci. U. S. A. 100 (14), 8053-8058 (1073).
- Ishihara, A., Hou, Y., Jacobson, K. The Thy-1 antigen exhibits rapid lateral diffusion in the plasma membrane of rodent lymphoid cells and fibroblasts. 84 (5), 1290-1293 (1987).
- Axelrod, D., et al. Lateral motion of fluorescently labeled acetylcholine receptors in membranes of developing muscle fibers. Proc. Natl. Acad. Sci. U. S. A. 73 (12), 4594-4598 (1976).
- Jacobson, K., Derzko, Z., Wu, E. S., Hou, Y., Poste, G. Measurement of the lateral mobility of cell surface components in single, living cells by fluorescence recovery after photobleaching. J. Supramol. Struct. 5 (4), 10-1002 (1976).
- Kusumi, A., et al. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34, 351-378 (2005).
- Kusumi, A., Ike, H., Nakada, C., Murase, K., Fujiwara, T. Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin. Immunol. 17 (1), 3-21 (2005).
- Schwille, P., Korlach, J., Webb, W. W. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry. 36, 176-182 (1999).
- Gielen, E., et al. Diffusion of sphingomyelin and myelin oligodendrocyte glycoprotein in the membrane of OLN-93 oligodendroglial cells studied by fluorescence correlation spectroscopy. C. R. Biol. 328 (12), 1057-1064 (2005).
- Weiss, M., Hashimoto, H., Nilsson, T. Anomalous protein diffusion in living cells as seen by fluorescence correlation spectroscopy. Biophys. J. 84, 4043-4052 (2003).
- Wawrezinieck, L., Rigneault, H., Marguet, D., Lenne, P. F. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys. J. 89 (6), 4029-4042 (2005).
- Lenne, P. F., et al. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 25 (14), 3245-3256 (2006).
- Ries, J., Schwille, P. Studying slow membrane dynamics with continuous wave scanning fluorescence correlation spectroscopy. Biophys. J. 91 (5), 1915-1924 (2006).
- Ruan, Q., Cheng, M. A., Levi, M., Gratton, E., Mantulin, W. W. Spatial-temporal studies of membrane dynamics: scanning fluorescence correlation spectroscopy (SFCS). Biophys. J. 87 (2), 1260-1267 (2004).
- Berland, K. M., So, P. T., Chen, Y., Mantulin, W. W., Gratton, E. Scanning two-photon fluctuation correlation spectroscopy: particle counting measurements for detection of molecular aggregation. Biophys. J. 71, 410-420 (1996).
- Heinemann, F., Betaneli, V., Thomas, F. A., Schwille, P. Quantifying lipid diffusion by fluorescence correlation spectroscopy: a critical treatise. Langmuir. 28 (37), 13395-13404 (2012).
- Cardarelli, F., Lanzano, L., Gratton, E. Capturing directed molecular motion in the nuclear pore complex of live cells. Proc. Natl. Acad. Sci. U. S. A. 109 (25), 9863-9868 (2012).
- Sanchez, S. A., Tricerri, M. A., Gratton, E. Laurdan generalized polarization fluctuations measures membrane packing micro-heterogeneity in vivo. Proc. Natl. Acad. Sci. U. S. A. 109 (19), 7314-7319 (2012).
- Cardarelli, F., Lanzano, L., Gratton, E. Fluorescence correlation spectroscopy of intact nuclear pore complexes. Biophys. J. 101 (4), 27-29 (2012).
- Di Rienzo, C., et al. Unveiling LOX-1 receptor interplay with nanotopography: mechanotransduction and atherosclerosis onset. Sci. Rep. 3, 10-1038 (2013).
- Unruh, J. R., Gratton, E. Analysis of molecular concentration and brightness from fluorescence fluctuation data with an electron multiplied CCD camera. Biophys. J. 95 (11), 5385-5398 (2008).
- Kannan, B., et al. Electron multiplying charge-coupled device camera based fluorescence correlation spectroscopy. Anal. Chem. 78 (10), 3444-3451 (2006).
- Jones, S. A., Shim, S. H., He, J., Fast Zhuang, X. three-dimensional super-resolution imaging of live cells. Nat. Methods. 8 (6), 499-508 (2011).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy. 3 (10), 793-795 (2006).
- Betzig, E., et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642-1645 (2006).
- Hess, S. T., Girirajan, T. P., Mason, M. D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91 (11), 4258-4272 (2006).
- Manley, S., et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods. 5 (2), 155-157 (2008).
- Hell, S. W. Far-field optical nanoscopy. Science. 316 (5828), 1153-1158 (2007).
- Klar, T. A., Hell, S. W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 24 (14), 954-956 (1999).
- Eggeling, C., et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 457 (7233), 1159-1162 (2009).
- Hedde, P. N., et al. Stimulated emission depletion-based raster image correlation spectroscopy reveals biomolecular dynamics in live cells. Nat. Commun. 4, Forthcoming.
- Hebert, B., Costantino, S., Wiseman, P. W. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 88 (5), 3601-3614 (2005).
- Brown, C. M., et al. Probing the integrin-actin linkage using high-resolution protein velocity mapping. J. Cell Sci. 119, 5204-5214 (2006).
- Di Rienzo, C., Gratton, E., Beltram, F., Cardarelli, F. Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. U. S. A. 110 (30), 12307-12312 (2013).
- Mueller, V., et al. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys. J. 101 (7), 1651-1660 (2011).
- Kleusch, C., Hersch, N., Hoffmann, B., Merkel, R., Csiszar, A. Fluorescent lipids: functional parts of fusogenic liposomes and tools for cell membrane labeling and visualization. Molecules. 17 (1), 1055-1073 (2012).
- Ries, J., Chiantia, S., Schwille, P. Accurate determination of membrane dynamics with line-scan FCS. Biophys. J. 96 (5), 1999-2008 (2009).
- Kolin, D. L., Wiseman, P. W. Advances in image correlation spectroscopy: measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cells. Cell Biochem. Biophys. 49 (3), 141-164 (2007).
- Digman, M. A., et al. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 89 (2), 1317-1327 (2005).
- Ritchie, K., et al. Detection of non-Brownian diffusion in the cell membrane in single molecule tracking. Biophys. J. 88 (3), 2266-2277 (2005).
- Voie, A. H., Burns, D. H., Spelman, F. A. Orthogonal-plane fluorescence optical sectioning: three-dimensional imaging of macroscopic biological specimens. J. Microsc. 170, 229-236 (1993).
- Huisken, J., Swoger, J., Del Bene,, Wittbrodt, F., J,, Stelzer, E. H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 305 (5686), 1007-1009 (2004).
- Wohland, T., Shi, X., Sankaran, J., Stelzer, E. H. Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments. Opt. Express. 18 (10), 10627-10641 (2010).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}