

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Este artículo describe los protocolos para la preparación de muestras, la reducción de datos y el análisis de datos en estudios de eco de espín de neutrones (NSE) de membranas lipídicas. El etiquetado juicioso de los lípidos con deuterio permite el acceso a diferentes dinámicas de membrana en escalas de longitud y tiempo mesoscópicas, sobre las cuales ocurren procesos biológicos vitales.
Abstract
Las bicapas lipídicas forman la matriz principal de las membranas celulares y son la plataforma principal para el intercambio de nutrientes, las interacciones proteína-membrana y la brotación viral, entre otros procesos celulares vitales. Para una actividad biológica eficiente, las membranas celulares deben ser lo suficientemente rígidas como para mantener la integridad de la célula y sus compartimentos, pero lo suficientemente fluidas como para permitir que los componentes de la membrana, como las proteínas y los dominios funcionales, se difundan e interactúen. Este delicado equilibrio de las propiedades de la membrana elástica y fluida, y su impacto en la función biológica, requieren una mejor comprensión de la dinámica colectiva de la membrana en escalas de tiempo y duración mesoscópicas de procesos biológicos clave, por ejemplo, deformaciones de membrana y eventos de unión a proteínas. Entre las técnicas que pueden sondear eficazmente este rango dinámico se encuentra la espectroscopia de eco de espín de neutrones (NSE). Combinado con el etiquetado de deuterio, NSE se puede utilizar para acceder directamente a las fluctuaciones de flexión y espesor, así como a la dinámica mesoscópica de características de membrana seleccionadas. Este documento proporciona una breve descripción de la técnica NSE y describe los procedimientos para realizar experimentos de NSE en membranas liposomales, incluidos los detalles de la preparación de muestras y los esquemas de deuteración, junto con instrucciones para la recopilación y reducción de datos. El documento también presenta métodos de análisis de datos utilizados para extraer parámetros clave de la membrana, como el módulo de rigidez de flexión, el módulo de compresibilidad del área y la viscosidad en el plano. Para ilustrar la importancia biológica de los estudios de NSE, se discuten ejemplos seleccionados de fenómenos de membrana sondeado por NSE, a saber, el efecto de los aditivos en la rigidez de flexión de la membrana, el impacto de la formación de dominios en las fluctuaciones de la membrana y la firma dinámica de las interacciones membrana-proteína.
Introduction
La comprensión de las membranas celulares y su función ha evolucionado notablemente en las últimas décadas. La visión anterior de las membranas celulares como bicapas lipídicas pasivas que definen los límites celulares y albergan proteínas de membrana1 se ha transformado gradualmente en un modelo dinámico en el que las bicapas lipídicas desempeñan un papel importante en la regulación de los procesos biológicos vitales, incluida la señalización celular, el intercambio molecular y la función de las proteínas, por nombrar algunos2,3,4,5,6. Esta comprensión de que las membranas celulares son altamente dinámicas, en constante remodelación y redistribución molecular, ha impulsado exploraciones científicas más allá de las estructuras de equilibrio de las membranas7,8,9. En consecuencia, se han desarrollado múltiples enfoques para estudiar los diversos modos dinámicos en las membranas lipídicas biológicas y bioinspiradas. Hasta la fecha, la mayoría de estos estudios se han centrado principalmente en los movimientos moleculares difusivos10,11,12,13 y las fluctuaciones macroscópicas de la forma14,15,16,dejando un vacío significativo en la comprensión de la dinámica intermedia de la membrana, es decir, las fluctuaciones colectivas de los conjuntos de lípidos que consisten en unos pocos 10-100 de moléculas de lípidos. Estas dinámicas ocurren en escalas de longitud de pocas decenas a pocas 100 Å y en escalas de tiempo de sub-ns a unos pocos cientos de ns (ver Figura 1),referidas aquí como escalas mesoscópicas. De hecho, es en estas escalas que la actividad biológica clave tiene lugar a nivel de membrana17. Esto incluye la brotación viral18,la canalización19y las interacciones membrana-proteína20. También es importante señalar que el panorama energético de las proteínas de membrana21,22 muestra que los cambios conformacionales en las proteínas, necesarios para su papel regulador, ocurren en las escalas de tiempons 23 de fluctuaciones colectivas de la membrana, enfatizando aún más la importancia de la dinámica mesoscópica en la función biológica de las membranas celulares y sus análogos bioinspirados20. Este artículo se centra en los dos modos dinámicos mesoscópicos primarios en las membranas lipídicas, a saber, las fluctuaciones de flexión y las fluctuaciones de espesor.
El principal desafío al sondear directamente estos modos de fluctuación es la dificultad de acceder simultáneamente a sus escalas espaciales y temporales utilizando métodos de espectroscopia estándar. El otro desafío es que las técnicas de contacto directo podrían afectar las mismas fluctuaciones que se supone que miden16. Esto se ve agravado por la complejidad composicional y estructural de las membranas biológicas24,25,lo que resulta en características de membrana no homogéneas, incluida la formación de dominios lipídicos26,27,28,29,30 y la asimetría de membrana31,32,33, que exigen sondas selectivas para comprender la dinámica de diferentes características de la membrana. Afortunadamente, estos desafíos pueden superarse con métodos de espectroscopia de neutrones no invasivos, como el eco de espín de neutrones (NSE), que inherentemente accede a las escalas de longitud y tiempo requeridas, y permiten además estudios de características selectivas de la membrana sin cambiar su entorno fisicoquímico34. De hecho, en los últimos años la espectroscopia NSE se ha convertido en una sonda única y poderosa de dinámica colectiva demembranas 35. Los resultados de los estudios de NSE sobre membranas lipídicas han producido nuevos conocimientos sobre las propiedades mecánicas36,37 y viscoelásticas38,39 de las membranas lipídicas y han arrojado nueva luz sobre su papel potencial en la función biológica40,41.
La técnica de espectroscopia NSE se basa en un diseño de instrumento interferométrico, propuesto por primera vez por Mezei42,utilizando una serie de aletas de espín y bobinas magnéticas para controlar la precesión del espín de neutrones a medida que los neutrones atraviesan el instrumento. El diseño se basa en el reflejo magnético de los elementos del campo magnético con respecto a la posición de la muestra (Figura 1A). Esto implica que en ausencia de intercambio de energía entre el neutrón y la muestra, el neutrón realiza el mismo número de precesiones de espín, en direcciones opuestas, en la primera y segunda mitad del instrumento (nótese el π-flipper entre las dos bobinas de precesión). Como resultado, el estado de espín final del neutrón permanece sin cambios en relación con el estado inicial, un fenómeno conocido como espín-eco (ver neutrón transparente en la Figura 1A). Sin embargo, cuando el neutrón interactúa energéticamente con la muestra, el intercambio de energía modifica el número de precesiones de espín en la segunda mitad del instrumento, lo que lleva a un estado de espín final diferente (ver Figura 1A). Esto se detecta experimentalmente como una pérdida en la polarización, como se mostrará más adelante en este artículo. Para obtener más detalles sobre la técnica NSE, se remite al lector a los documentos técnicos dedicados42,43,44,45.
Aquí, se presenta una descripción simplificada para proporcionar una estimación aproximada de la longitud y las escalas de tiempo accesibles con NSE. Las escalas de longitud están determinadas por el rango de transferencias de vector de onda alcanzables, Q = 4π sin θ/λ, donde 2θ es el ángulo de dispersión y λ es la longitud de onda del neutrón. Se puede ver que Q está establecido por el rango de longitud de onda y la extensión de rotación del segundo brazo del espectrómetro (ver Figura 1A). Un rango Qtípico en los espectrómetros NSE es ~ 0.02-2 Å-146,47y hasta 0.01-4 Å-1 con actualizaciones recientes48,49, correspondientes a escalas espaciales de ~ 1-600 Å. Por otro lado, la escala de tiempo accesible se calcula a partir del ángulo de precesión total (o fase) adquirido por el neutrón dentro de las bobinas de precesión magnética, y se encuentra que esde 50:  . En esta expresión, t es el tiempo de Fourier definido como
. En esta expresión, t es el tiempo de Fourier definido como  , donde es la relación
, donde es la relación  giromagnética de neutrones,
giromagnética de neutrones,  es la longitud de la bobina y es la fuerza del campo magnético de
es la longitud de la bobina y es la fuerza del campo magnético de  la bobina. Vale la pena señalar que el tiempo de Fourier es una cantidad que depende estrictamente de la geometría del instrumento, la intensidad del campo magnético y la longitud de onda de los neutrones. Por ejemplo, utilizando neutrones de longitud de onda
la bobina. Vale la pena señalar que el tiempo de Fourier es una cantidad que depende estrictamente de la geometría del instrumento, la intensidad del campo magnético y la longitud de onda de los neutrones. Por ejemplo, utilizando neutrones de longitud de onda  = 8 Å y ajustes del instrumento de = 1,2 m y = 0,4 T, el tiempo de Fourier se calcula en t ~ 50 ns. Experimentalmente, el tiempo de Fourier se ajusta cambiando la corriente en las bobinas de precesión (es decir, la intensidad del campo magnético) o utilizando diferentes longitudes de onda de neutrones, lo que resulta en escalas de tiempo típicas de NSE de ~ 1 ps a 100 ns. Sin embargo, las actualizaciones recientes en los espectrómetros NSE han permitido el acceso a tiempos Fourier más largos, hasta ~ 400 ns en el espectrómetro J-NSE-Phoenix en el Heinz Maier-Leibnitz Zentrum51 y el espectrómetro SNS-NSE en el Laboratorio Nacional Oak Ridge48,y hasta ~ 1,000 ns en el espectrómetro IN15 NSE en el Institut Laue-Langevin (ILL)49.
= 8 Å y ajustes del instrumento de = 1,2 m y = 0,4 T, el tiempo de Fourier se calcula en t ~ 50 ns. Experimentalmente, el tiempo de Fourier se ajusta cambiando la corriente en las bobinas de precesión (es decir, la intensidad del campo magnético) o utilizando diferentes longitudes de onda de neutrones, lo que resulta en escalas de tiempo típicas de NSE de ~ 1 ps a 100 ns. Sin embargo, las actualizaciones recientes en los espectrómetros NSE han permitido el acceso a tiempos Fourier más largos, hasta ~ 400 ns en el espectrómetro J-NSE-Phoenix en el Heinz Maier-Leibnitz Zentrum51 y el espectrómetro SNS-NSE en el Laboratorio Nacional Oak Ridge48,y hasta ~ 1,000 ns en el espectrómetro IN15 NSE en el Institut Laue-Langevin (ILL)49.
Además del acceso directo a la escala de tiempo y longitud de la dinámica de membranas, NSE tiene las capacidades inherentes de la sensibilidad a los isótopos de neutrones52. Específicamente, la capacidad de los neutrones para interactuar de manera diferente con los isótopos del hidrógeno, el elemento más abundante en los sistemas biológicos, da como resultado una densidad de longitud de dispersión de neutrones diferente,34 o NSLD (el equivalente al índice óptico de refracción50),cuando el protio es sustituido por deuterio. Esto permite un enfoque conocido como variación de contraste, que se usa comúnmente para resaltar características específicas de la membrana u ocultar otras; este último escenario se conoce como coincidencia de contraste. Una aplicación frecuente de variación/coincidencia de contraste es la sustitución de agua (NSLD = -0.56 × 10-6 Å-2) por agua pesada o D2O (NSLD = 6.4 × 10-6 Å -2) para amplificar la señal de neutrones de las membranas lipídicas protiatadas (NSLD ~ 0 × 10-6 Å-2). Este enfoque es altamente efectivo en estudios de estructura de membrana porque la penetración de D2O en la región del grupo de cabeza de la membrana permite la determinación precisa de los espesores de la membrana (ver Figura 2A,panel izquierdo) y de la ubicación de diferentes subgrupos lipídicos cuando se aplican modelos más sofisticados53,54. Este artículo destaca algunos ejemplos sobre el uso de la variación de contraste para estudios de dinámica colectiva en membranas biomiméticas y características de membrana seleccionadas.
Aquí, la efectividad de NSE para proporcionar información única sobre las propiedades dinámicas y funcionales de la membrana se ilustra a través de ejemplos tangibles de estudios de NSE en sistemas de membrana lipídica modelo y biológicamente relevantes con énfasis en la dinámica de mesoescala en membranas independientes, en forma de suspensiones liposomales. Para las mediciones de NSE de la dinámica de la membrana en el plano, se remite al lector a publicaciones dedicadas a la espectroscopia de espín-eco de neutrones de incidencia de pastoreo (GINSES)55,56 y otros estudios de pilas de membrana multilamelar alineadas57,58,59,60.
Para simplificar, este artículo destaca tres esquemas diferentes de deuteración de membrana ilustrados en un sistema de bicapa lipídica de formación de dominios o separación de fases bien estudiado de mezclas de 1,2-ditiristoil-sn-glicero-3-fosfocolina (DMPC) y 1,2-distearoil-sn-glicero-3-fosfocolina (DSPC)61,62. Los dos lípidos se caracterizan por un desajuste en la longitud de su cadena de hidrocarburos (14 carbonos/cola en DMPC vs 18 carbonos/cola en DSPC) y su temperatura de transición gel-fluido (Tm, DMPC = 23 °C vs Tm, DSPC = 55 °C). Esto da como resultado la separación de fases laterales en membranas DMPC:DSPC a temperaturas entre las temperaturas de transición superior e inferior de la mezcla63. Los esquemas de deuteración considerados aquí se eligen para demostrar los diferentes modos dinámicos accesibles en las mediciones de NSE en membranas liposomales, a saber, fluctuaciones de flexión, fluctuaciones de espesor y fluctuaciones selectivas de flexión / espesor de dominios laterales. Todas las composiciones lipídicas se informan para las bicapas DMPC:DSPC preparadas a una fracción molar de 70:30, utilizando variantes protiated y perdeuteradas disponibles comercialmente de DMPC y DSPC. Todos los pasos de preparación de la muestra se basan en 4 mL de suspensión liposomal, en D2O, con una concentración de lípidos de 50 mg/mL, para una masa lipídica total de Mtot = 200 mg por muestra.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Esquema de deuteración requerido para el experimento
- Para las mediciones de fluctuación de flexión, realice liposomas completamente protiated en D2O (D 99.9%) o D2O-buffer (por ejemplo, tampón de fosfato preparado con D2O en lugar de H2O). Utilice DMPC (C36H72NO8P) y DSPC (C44H88NO8P) totalmente protiados con
 133,4 mg, donde XDMPC y XDSPC son las fracciones molares de DMPC y DSPC, aquí establecidas en 0,7 y 0,3, respectivamente, y MwDMPC y MwDSPC son los pesos molares dados por 677,9 g/mol y 790,1 g/mol, respectivamente. Del mismo modo, mDSPC = 66,6 mg. Este esquema de deuteración aumenta el contraste de dispersión entre la membrana (NSLD ~ 0 × 10-6 Å -2) y el tampón deuterado (NSLD ~ 6.4 × 10-6 Å -2) y amplifica la señal de las ondulaciones de membrana (ver Figura 2A panel izquierdo).
133,4 mg, donde XDMPC y XDSPC son las fracciones molares de DMPC y DSPC, aquí establecidas en 0,7 y 0,3, respectivamente, y MwDMPC y MwDSPC son los pesos molares dados por 677,9 g/mol y 790,1 g/mol, respectivamente. Del mismo modo, mDSPC = 66,6 mg. Este esquema de deuteración aumenta el contraste de dispersión entre la membrana (NSLD ~ 0 × 10-6 Å -2) y el tampón deuterado (NSLD ~ 6.4 × 10-6 Å -2) y amplifica la señal de las ondulaciones de membrana (ver Figura 2A panel izquierdo). - Para medir la dinámica de flexión de características seleccionadas de la membrana lateral, por ejemplo, dinámica de la matriz en membranas DMPC:DSPC de separación de fase, use DMPC protiatado (C36H72NO8P) y deuterado, DSPC-d83 (C44H5NO8PD83, Mw 873.7 g / mol), de modo que mDMPC = 128.8 mg y mDSPC-d83 = 71.2 mg. Este esquema de deuteración minimiza la dispersión de los dominios ricos en DSPC no deseados, lo que permite mediciones selectivas de las fluctuaciones de flexión de la matriz rica en DMPC (consulte la Figura 2B del medio).
NOTA: Para encontrar la deuteración lipídica óptima requerida para un esquema específico de coincidencia de contraste, utilice calculadoras de densidad de longitud de dispersión (SLD) disponibles basadas en la web, como la desarrollada por el Centro NIST para la Investigación de Neutrones64. Estas interfaces basadas en la web están equipadas con herramientas fáciles de usar para facilitar el cálculo del SLD de lípidos con varios grados de deuteración, así como el de las mezclas de lípidos. - Para las mediciones de NSE de las fluctuaciones medias del espesor de la membrana (sin contraste lateral), utilice variantes deuteradas de la cola de los lípidos constituyentes, es decir, DMPC-d54 (C36H18NO8PD54, 732,3 g/mol) y DSPC-d70 (C44H18NO8PD70, 860,1 g/mol)35,38, tal que mDMPC-d54 = 133,0 mg y mDSPC-d70 = 67,0 mg. Este esquema de contraste(Figura 2A,panel derecho) amplifica la señal de dispersión de los grupos de cabeza de lípidos (NSLD ~ 4.5 × 10-6 Å -2) haciendo coincidir el grupo de cola (NSLD ~ 6.4 × 10-6 Å -2) con el tampón deuterado que permite la detección de fluctuaciones en el espesor de la membrana.
- Para los estudios de fluctuación de espesor de compartimentos de membrana seleccionados, por ejemplo, matriz rica en DMPC, utilice la misma estrategia descrita en el paso 1.2 sustituyendo los lípidos DMPC protiados con sus análogos deuterados de cola, es decir, DMPC-d54, de modo que los dominios ricos en DSPC se emparejen con contraste con el tampón deuterado y la señal de dispersión primaria provenga de la región del grupo de cabeza de la matriz rica en DMPC deuterada en cola.
 133,4 mg, donde XDMPC y XDSPC son las fracciones molares de DMPC y DSPC, aquí establecidas en 0,7 y 0,3, respectivamente, y MwDMPC y MwDSPC son los pesos molares dados por 677,9 g/mol y 790,1 g/mol, respectivamente. Del mismo modo, mDSPC = 66,6 mg. Este esquema de deuteración aumenta el contraste de dispersión entre la membrana (NSLD ~ 0 × 10-6 Å -2) y el tampón deuterado (NSLD ~ 6.4 × 10-6 Å -2) y amplifica la señal de las ondulaciones de membrana (ver Figura 2A panel izquierdo).
133,4 mg, donde XDMPC y XDSPC son las fracciones molares de DMPC y DSPC, aquí establecidas en 0,7 y 0,3, respectivamente, y MwDMPC y MwDSPC son los pesos molares dados por 677,9 g/mol y 790,1 g/mol, respectivamente. Del mismo modo, mDSPC = 66,6 mg. Este esquema de deuteración aumenta el contraste de dispersión entre la membrana (NSLD ~ 0 × 10-6 Å -2) y el tampón deuterado (NSLD ~ 6.4 × 10-6 Å -2) y amplifica la señal de las ondulaciones de membrana (ver Figura 2A panel izquierdo).2. Preparación de suspensión lipídica para extrusión
- Calcule la masa de cada componente de la muestra, dependiendo de la composición de la muestra. Como regla general, para muestras con múltiples componentes moleculares, la masa de un componente está dada por su masa molar, Mwi,ponderada por su fracción molar, Xi,y normalizada sobre todos los componentes de tal manera que:
 donde Mtot es la masa total, establecida aquí en 200 mg. Vea el ejemplo anterior para las bicapas lipídicas DMPC-DSPC con diferentes esquemas de deuteración.
donde Mtot es la masa total, establecida aquí en 200 mg. Vea el ejemplo anterior para las bicapas lipídicas DMPC-DSPC con diferentes esquemas de deuteración. - Usando una semimicrapalanza digital, sopese las masas calculadas de lípidos (y otros componentes de la muestra, por ejemplo, proteínas, nanopartículas, etc.) y agréguelas a un vial o matraz de fondo redondo; recuerde pesar el vial o matraz de antemano. Agregue 1 ml de disolvente para disolver los componentes pesados mezclando manualmente dentro de una campana. Para muestras de lípidos puros, use cloroformo o etanol. Para muestras con componentes adicionales no lipídicos (por ejemplo, nanopartículas), elija un disolvente común que disperse todos los componentes.
- Para pequeñas cantidades de lípidos (<10 mg), prepare una solución de caldo y pipetee el volumen requerido en la mezcla.
NOTA: No agregue cantidades excesivas de solvente, ya que ralentizará significativamente el paso de secado con solvente que se describe a continuación.
- Para pequeñas cantidades de lípidos (<10 mg), prepare una solución de caldo y pipetee el volumen requerido en la mezcla.
- Seque la solución lipídica, dentro de una campana, transmitiendo suavemente un gas inerte (por ejemplo, nitrógeno, argón) en el vial mientras gira lentamente el vial en ángulo. Mantenga los viales en posición inclinada para crear una película delgada de lípidos secos en las paredes del vial, lo que permitirá un secado uniforme. Coloque intermitentemente el vial en un baño de agua a 35 °C para evitar el enfriamiento mediado por evaporación, lo que ralentizará la evaporación del disolvente.
- Coloque los viales durante la noche en un horno al vacío a ~ 35 ° C para eliminar completamente el disolvente residual. Para los lípidos insaturados, purgue el vacío con un gas inerte para minimizar la oxidación.
- Para garantizar la eliminación completa del disolvente, pese el vial después del secado de lípidos y confirme que no hay exceso de masa más allá de las cantidades medidas de materiales. Haga esto restando la masa del vial de la masa medida después del secado. Si hay exceso de masa, seque la muestra al vacío durante otras 6 h. Repita este proceso según sea necesario.
- Hidratar la película lipídica con 4 mL de D2O o D2O-buffer para obtener una concentración lipídica de 50 mg/mL. Para lípidos con altas temperaturas de transición, como las mezclas DMPC-DSPC, caliente el tampón por encima de la temperatura de transición (60 ° C) para garantizar una mezcla uniforme.
NOTA: Dado que los experimentos con NSE requieren volúmenes de muestra relativamente grandes (~4 ml), considere la posibilidad de hidratar la muestra utilizando la mitad del tampón requerido, es decir, 2 ml, para minimizar el número de extrusiones por muestra (ver sección 3). En este caso, agregue la mitad restante del búfer después de la extrusión. Tenga en cuenta que la capacidad de las jeringas utilizadas en la extrusión está limitada a 1 ml. Por lo tanto, hidratarse con 4 ml de tampón requeriría cuatro series de extrusión. - Mezcle con vórtice la solución lipídica hidratada hasta que la película lipídica se disuelva por completo y ya no sea visible en las paredes del vial. En esta etapa, los lípidos hidratados forman vesículas multilamelares y pilas multilamelares de tamaño micrométrico y la suspensión aparece de color blanco lechoso.
- Para facilitar la rotura de las pilas de lípidos y reducir la multilamelaridad, realice cinco ciclos de congelación/descongelación colocando el vial de solución lipídica hidratada en un congelador de grado de laboratorio (preferiblemente congelador de -80 °C) hasta que esté completamente congelado y luego transfiriendo el vial a un baño de agua de 35 °C hasta que la solución lipídica se descongele por completo. Vórtice la solución descongelada hasta que sea homogénea. Repetir cuatro veces más.
NOTA: Alternativamente, se puede preparar un baño de hielo seco para una congelación rápida combinando acetona y hielo seco.
 donde Mtot es la masa total, establecida aquí en 200 mg. Vea el ejemplo anterior para las bicapas lipídicas DMPC-DSPC con diferentes esquemas de deuteración.
donde Mtot es la masa total, establecida aquí en 200 mg. Vea el ejemplo anterior para las bicapas lipídicas DMPC-DSPC con diferentes esquemas de deuteración.3. Extrusión de la solución lipídica hidratada
- Ensamble la configuración de la extrusora utilizando una membrana de policarbonato entre dos soportes de membrana y agregando dos filtros de papel en cada lado para proporcionar un soporte adicional. Use una membrana de policarbonato con un tamaño de poro que coincida con el tamaño liposomal objetivo (los tamaños de poro comunes para los experimentos de NSE son 50 nm y 100 nm; por lo general, los liposomas de 100 nm de diámetro permiten fluctuaciones de membrana menos restringidas, pero se podrían usar liposomas más pequeños de 50 nm para estudios de curvatura). Asegúrese de que la membrana de policarbonato esté completamente estirada antes de completar el ensamblaje y apretar la carcasa externa del extrusor.
- Hidrate la membrana de policarbonato pasando ~ 0.3 mL de D2O o D2O-buffer varias veces a través del conjunto de membrana usando jeringas de vidrio herméticas. Utilice el mismo búfer utilizado en la preparación de muestras. Déjelo durante al menos 10 minutos, luego succione completamente el búfer antes de introducir la muestra.
- Llene una jeringa hermética al gas de 1 ml con la solución lipídica preparada e insértela en un extremo del aparato extrusor. Luego, inserte una jeringa vacía en el extremo opuesto. Una vez que las jeringas estén conectadas al conjunto de la extrusora, colóquela en el bloque de la extrusora.
- Si se necesitan temperaturas elevadas para la extrusión, como en el caso de los lípidos saturados con altas temperaturas de transición (por ejemplo, DSPC, Tm = 55 °C), precaliente el bloque de calentamiento del extrusor por encima de la temperatura de transición lipídica (por ejemplo, 60 °C), colocando el bloque calefactor en una placa caliente o utilizando un baño de circulación como se muestra en la Figura 3A.
NOTA: Este paso es crucial para garantizar una mezcla homogénea de lípidos y para evitar ejercer una presión extrema durante la extrusión, lo que podría romper la membrana de policarbonato. Para muestras de lípidos con bajas temperaturas de transición (<25 °C), realice la extrusión a temperatura ambiente. - Para extruir la solución lipídica, conecte el conjunto de extrusoras a una bomba de jeringa programable con un marco de aluminio/acero como se muestra en la Figura 3A. Para extrusiones con temperatura controlada, agregue una base de extrusora personalizada con un canal de fluido y conéctela a un baño de agua circulante.
- Programe la bomba de jeringa para realizar 15-20 ciclos de extrusión siguiendo el manual del fabricante. Cuando se extruye, el color de la solución lipídica cambia de blanco lechoso a azul ópalo transparente(Figura 3B,C),lo que indica un tamaño liposomal final que es más pequeño que la longitud de onda de la luz visible, como se esperaba. Para el tipo de bomba de jeringa que se muestra en la Figura 3A,siga los pasos a continuación.
- Comience ajustando la configuración de la bomba. Mantenga pulsado el botón Velocidad e introduzca la velocidad de extrusión (50,99 ml/h), pulse el botón Diámetro e introduzca el diámetro de la jeringa (4,606 mm). Use las flechas hacia arriba debajo de cada dígito en la pantalla para cambiar ese valor de dígito.
- Coloque el juego de extrusoras con la jeringa de muestra a la derecha (consulte la Figura 3A). Pulse el botón Retirar hasta que se encienda la luz de retirada. Presione Inicio y espere a que la muestra se dispense en la jeringa izquierda (vacía).
- Presione el botón Detener justo antes de que la jeringa de muestra (derecha) esté completamente vacía. Registre el volumen dispensado y utilítelo para programar el ciclo de extrusión. Mantenga pulsado el botón Tasa hasta que aparezca la fase 1 (PH:01) en la pantalla. Pulse el botón Volumen para introducir el volumen dispensado registrado anteriormente. En esta fase, asegúrese de que la luz De extracción esté apagada, lo que dispensa la muestra en la dirección correcta.
- Pulse de nuevo el botón Tasa y utilice la flecha hacia arriba más a la derecha para acceder a la fase 2 (PH:02). Pulse Volumen para introducir el mismo valor del volumen dispensado registrado anteriormente. En esta fase, presione el botón Retirar hasta que la luz Retirar esté encendida, lo que dispensa la muestra hacia la izquierda.
- Para repetir este ciclo, presione el botón Tasa nuevamente y use la flecha hacia arriba más a la derecha para acceder a la fase 3 (PH: 03). Presione el botón Volumen hasta que LP: SE aparezca en la pantalla y configátelo en 20. Este es el número de bucles o repeticiones que realizará la bomba. Finalmente, presione el botón Tasa, acceda a la fase 4 (PH: 04) y presione el botón Volumen para llegar a la función Detener. La bomba ahora está configurada para la extrusión automatizada.
- Pulse Inicio para iniciar el ciclo de extrusión.
- Vacíe la jeringa que contiene la suspensión de lípidos extruidos en un vial limpio y prepárela para el almacenamiento o las mediciones. Para muestras de lípidos con alta temperatura de fusión, almacene la muestra por encima de la transición de fase fluida hasta que se mida. De lo contrario, mantenga las muestras a temperatura ambiente.
- No congele las muestras extruidas, ya que la congelación hará que las vesículas se rompan (la suspensión volverá a ser de color blanco lechoso).
4. Mediciones de NSE para la(s) muestra(s) y reducción de los datos recogidos
- Antes del experimento NSE, caracterizar la muestra liposomal extruida del paso 3.7 utilizando los métodos disponibles para garantizar una calidad de muestra adecuada. En la sección de discusión se incluye una lista de posibles métodos de charcaterización que se pueden utilizar para evaluar la calidad de las suspensiones liposomales para experimentos de NSE, por ejemplo, distribución de tamaño, multilamelaridad, estructura de membrana lateral.
- Determine el rango Q y la configuración del instrumento correspondiente requerida para el experimento. Para las mediciones de rigidez de flexión de bicapas lipídicas, use un rango Q de ~(0.04 - 0.2) Å-1. Para estudios de fluctuaciones de espesor de membrana, use un rango Q de ~(0.04 - 0.2) Å-1 correspondiente al espesor de membrana35,66,67.
NOTA: Discuta la configuración experimental con el científico del instrumento antes del inicio del experimento. Como se mencionó anteriormente, la caracterización SANS de la muestra es necesaria, especialmente si no se dispone de información previa de la señal de dispersión, como en las membranas deuteradas selectivamente. Alternativamente, ejecute mediciones estáticas (también conocidas como difracción) en un rango Q limitado en el instrumento NSE, con la advertencia de que tales mediciones toman mucho más tiempo en comparación con SANS. - Usando una jeringa o una pipeta de transferencia, cargue la(s) suspensión(es) liposomal(es) extruida(s) en las células de muestra designadas disponibles en las líneas de haz de NSE. Tenga en cuenta que las células de muestra NSE estándar vienen en espesores de 1, 2, 3 y 4 mm. Elija el grosor de la celda de tal manera que optimice la señal de dispersión mientras mantiene la señal de fondo incoherente a una intensidad razonable.
NOTA: Como regla general, use células de muestra con una longitud de ruta de 1 o 2 mm para liposomas protiados en tampón deuterado: las células más gruesas podrían provocar múltiples efectos de dispersión que son difíciles de corregir. Para los liposomas con niveles más altos de deuteraciones (por ejemplo, liposomas con contraste de cola o liposomas asimétricos con valvas protiadas únicas), considere el uso de una célula de muestra más gruesa (por ejemplo, longitud de ruta de 3 o 4 mm) para mejorar las estadísticas de conteo si la muestra está disponible en cantidades más grandes, a veces esto puede ser prohibitivo. - Prepare una celda de muestra idéntica para el búfer. Use el mismo tampón que en la suspensión liposomal. Las mediciones en el búfer son necesarias para la normalización de la intensidad y las correcciones de fondo (BKG).
- Coloque la(s) celda(s) de muestra en el soporte de muestra del espectrómetro NSE, programe las ejecuciones de medición y recopile datos de eco. Consulte con el científico del instrumento sobre la programación de las mediciones si es la primera vez que se usuario de NSE.
- Realice dos conjuntos adicionales de mediciones necesarias para la reducción de datos: mediciones de resolución (R) y transmisión (T).
- Realizar la medición de resolución(R)en una referencia de dispersión elástica (por ejemplo, carbono), que se ejecutará bajo la misma configuración; es decir, el mismo vector de onda y las mismas veces que las mediciones de muestra y tampón.
- Realizar mediciones de transmisión(T)en la muestra y el búfer para calcular la intensidad del haz de neutrones transmitido (véase el paso 4.9. a continuación). La transmisión se calcula como la relación de recuentos de neutrones de la muestra o tampón dividida por los recuentos de neutrones para un haz abierto (es decir, con una posición de muestra vacía).
- Utilice el software de reducción de datos dedicado para el espectrómetro NSE en el que se realizan las mediciones para reducir los datos recopilados.
NOTA: Diferentes espectrómetros pueden utilizar diferentes software o interfaces de usuario. A continuación se muestra un ejemplo de reducción de datos NSE utilizando el Entorno de Análisis y Visualización de Datos (DAVE)68 software escrito específicamente para el espectrómetro NSE en el Centro de Investigación de Neutrones del NIST.- Abra el software DAVE y seleccione Reducir datos NSE en el menú de reducción de datos. Aparecerán varias ventanas emergentes.
- Cargue los archivos de datos sobre diferentes valores Q utilizando el menú Abrir archivos .echo del menú archivo. Estos archivos corresponden a los archivos de datos sin procesar con las señales de eco de giro y tienen la extensión .echo en el nombre del archivo. Una vez que se complete la carga de archivos, los archivos se mostrarán debajo de los conjuntos de datos disponibles.
- Haga clic derecho en el archivo seleccionado y etiquételo de acuerdo con la medida a la que corresponde; es decir, Muestra, Celda (para celda vacía o búfer) o Resolución.
- Agrupo los pixles del detector en 2 x 2 para mejorar la relación señal-ruido utilizando la pestaña Conjunto de datos. Aplicar el mismo binning a todos los archivos; es decir, Resolución, Celda y Muestra.
- Inspeccione los datos en todos los grupos de píxeles y enmascare aquellos con señales deficientes (consulte la Figura 4B)presionando la tecla m en el teclado. Presione Entrar para acceder a una ventana emergente para aplicar la misma máscara a todas las veces de Fourier o posteriores de Fourier. Esto también se puede aplicar a píxeles individuales en cualquier punto durante la reducción de datos. Los píxeles enmascarados se volverán verdes.
- Asegúrese de que los datos recopilados estén en forma de una señal de eco, es decir, una función de coseno en términos de la corriente de fase, sobre cada píxel del detector (consulte la Figura 4A).
NOTA: La corriente de fase es proporcional al ángulo de precesión del espín de neutrones; por lo tanto, es común representar la corriente de fase como un ángulo de fase como se muestra en la Figura 4A. Para las mediciones en fuentes pulsadas, se aplican cálculos adicionales de tiempo de vuelo a los datos para obtener las señales de eco en función de la longitud de onda de neutrones incidentes dentro de un pulso de neutrones. - Comience por ajustar el archivo de resolución. Seleccione un archivo de resolución de la lista de archivos cargados y haga clic con el botón derecho en el archivo. Seleccione Operaciones de ajuste: Ajustar ecos (resolución) en el menú emergente.
- Asegúrese de que los ajustes de las señales de eco producen una serie de parámetros de ajuste, incluido el parámetro A, requerido en el paso 4.8. Los ajustes se realizan automáticamente utilizando la siguiente expresión.

Aquí, ζ es el período de la señal de eco (es decir, la función del coseno en la Figura 4A),σ es el ancho de la envoltura gaussiana determinado por la longitud de onda media y la extensión de la longitud de onda del haz de neutrones incidente, Φc es la corriente de fase y Φ0 es el punto de eco que depende de la trayectoria del campo experimentado por los neutrones50. La información física sobre la muestra está codificada en la amplitud, A, de la función coseno en la ecuación (1).
NOTA: El ancho de la envolvente gaussiana se basa en valores predeterminados por el científico del instrumento y no debe cambiarse. Los otros parámetros son variables que se ajustan a la señal de eco específica sobre cada píxel. - Inspeccione los resultados de ajuste haciendo clic en cada píxel para mostrar los parámetros de ajuste resultantes, la calidad del ajuste y la desviación cuadrada media del ajuste. Para inspeccionar el error asociado con cada parámetro de conexión en todo el detector, seleccione Opciones de imagen y, a continuación, seleccione el parámetro de conexión de interés. Esto generará un mapa con el valor del parámetro de ajuste sobre cada píxel. Haga clic con el botón derecho en la imagen del detector. Aparecerá una ventana emergente que muestra un mapa de barra de error del parámetro de conexión seleccionado.
- Si el ajuste sobre un píxel específico no es satisfactorio (por ejemplo, ajustar parameeters con barras de error grandes), reajuste la señal sobre ese píxel específico. Seleccione ese píxel, presione la ficha Conexión y, a continuación, presione Ajustar píxel. Introduzca nuevos parámetros de partida para la fase (Φ0) y el período (ζ) en la ficha Fitting para obtener un ajuste más satisfactorio.
NOTA: Es útil trazar la fase ajustada en función del tiempo de Fourier. Para hacerlo, vaya a la ventana principal de la trama y seleccione Fit Phase v. Fourier Time. Esta trama debe ser lisa y continua. Inspeccione las discontinuidades en esta gráfica y reajuste los píxeles a los que corresponden.
- Reduzca el archivo de muestra o celda seleccionando el archivo correspondiente de la lista de archivos cargados y etiquetados.
- Inspeccione todos los píxeles y enmascare los que tienen estadísticas incorrectas como se describe en el paso 4.7.5.
- Haga clic con el botón derecho en el archivo y seleccione Operaciones de ajuste: Importar fases (muestra, celda). Esto importa las fases y la máscara aplicada desde el archivo de resolución.
- Ajuste las señales de eco utilizando el mismo procedimiento descrito anteriormente para el archivo de resolución (pasos 4.7.8-4.7.10). Al ajustar los archivos de muestra y celda, no cambie los valores del punto de fase de punto y fase de eco importados desde los ajustes de resolución. Estos parámetros dependen de la configuración instrumental y no deben variar con las muestras.
- Antes de proceder a la reducción de datos, ingrese el centro de haz para todos los archivos de datos. Seleccione el archivo de datos, vaya a la pestaña General e introduzca los valores del centro de haz X e Y. Estos valores se registran durante el experimento.
- Una vez que se completen los ajustes a los archivos de muestra, celda y resolución, calcule la función de dispersión intermedia normalizada que se utilizará más adelante en el análisis e interpretación de datos. Para hacer eso, haga clic derecho en el archivo de muestra que se reducirá de la lista de archivos ajustados y seleccione Calcular I (Q) en el menú emergente. Aparecerá una ventana con opciones de entrada para los archivos Resolución y Celda (es decir, búfer) y el número de Q-arcs (consulte el paso 4.9). Después de ingresar toda la información requerida, presione el botón OK. Los resultados aparecerán en una nueva ventana.
NOTA: La reducción de datos se realiza de acuerdo con la siguiente ecuación para obtener la función de dispersión intermedia normalizada69.
donde t es el tiempo de Fourier, Narriba y Nabajo son los recuentos de neutrones en las configuraciones no spin-flip y spin-flip (medidas con las π/2-flippers apagados y el π-flipper apagado y encendido, respectivamente), y los superíndices, BKG y R,corresponden a las mediciones de fondo y resolución, respectivamente, como se define en los pasos 4.4 y 4.6. Nótese que la polarización del haz, por lo tanto, los cambios en el estado de espín debido al intercambio de energía entre el neutrón y la muestra se detecta como una caída en la polarización (desde la unidad).
haz, por lo tanto, los cambios en el estado de espín debido al intercambio de energía entre el neutrón y la muestra se detecta como una caída en la polarización (desde la unidad).
- Finalmente, agrupo los píxeles del detector en Q-arcos como se muestra en la Figura 4B para obtener la Q-dependenciade la función de dispersión intermedia normalizada, S(Q,t) / S(Q,0). Esto se conoce técnicamente como agrupación de datos y debe hacerse juiciosamente, es decir, teniendo en cuenta las estadísticas de conteo de la muestra y la desviación estándar esperada de los datos sobre los píxeles agrupados.
- Para muestras de dispersión fuerte, divida el detector en más arcos Q mientras mantiene barras de error razonables en la función de dispersión intermedia resultante, S(Q,t) / S(Q,0). Esto produce más puntos de datos Q y es importante para el procedimiento de análisis de datos que se describe a continuación. Tenga en cuenta que para muestras de dispersión débil, el binning excesivo da como resultado señales de desintegración deficientes, es decir, grandes barras de error en S(Q,t) / S(Q, 0 ), lo que podría resultar en grandes incertidumbres.


 haz, por lo tanto, los cambios en el estado de espín debido al intercambio de energía entre el neutrón y la muestra se detecta como una caída en la polarización (desde la unidad).
haz, por lo tanto, los cambios en el estado de espín debido al intercambio de energía entre el neutrón y la muestra se detecta como una caída en la polarización (desde la unidad).5. Análisis e interpretación de datos
- Ajustar las funciones de dispersión intermedia normalizadas, S(Q,t) / S(Q,0 ), obtenidas de la reducción de datos anterior a una función exponencial estirada con un exponente de estiramiento de 2/370.

NOTA: Un ejemplo de estos ajustes se proporciona en la Figura 5B. Los ajustes de S(Q,t) / S(Q,0) a la ecuación (3) producen las tasas de relajación dependientes de Q Γ(Q). - Trazar Γ(Q) en función de Q y ajustarse a un modelo adecuado para extraer los parámetros relevantes de la membrana.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Los estudios de NSE que acceden a las fluctuaciones de flexión se realizan típicamente en un rango Q de ~ (0.04 - 0.2) Å-1. Este rango Q corresponde a escalas de longitud intermedia entre el grosor de la membrana y el radio liposomal, donde domina la dinámica de flexión. La medición en un rango Q extendido puede dar acceso a modos dinámicos adicionales, incluida la difusión liposomal y la dinámica intramembrana. Para obtener más detalles sobre el cruce en la dinámica de membranas a la que accede NSE, consulte estas publicaciones relevantes25,71. Es importante destacar que las señales NSE son proporcionales a:  , donde Icoh e Iinc son, respectivamente, la intensidad de dispersión coherente e incoherente de la muestra. Por lo tanto, es aconsejable preparar muestras liposomales de NSE en tampones deuterados (es decir, tampones preparados con D2O en lugar de H2O) para minimizar la señal de dispersión incoherente, principalmente aportada por el contenido de hidrógeno de la muestra. Sin embargo, en algunos casos pueden ser necesarios esquemas intermedios de deuteración (es decir, utilizando mezclas de D2O y H2O) para obtener condiciones óptimas de contraste. Por lo general, las mediciones de NSE de las fluctuaciones de flexión de la membrana se realizan en liposomas totalmente protiated en tampón deuterado, conocidos como liposomas totalmente contrastados en la Figura 5. Este esquema de deuteración da como resultado una gran diferencia de NSLD entre el núcleo de membrana (~0 × 10-6 Å-2) y su entorno de fluido deuterado (~6.4 × 10-6 Å-2), lo que mejora significativamente la señal de dispersión de las membranas liposomales y mejora las estadísticas de medición de la dinámica de flexión. Este esquema decontraste(Figura 2A panel izquierdo) se utiliza con frecuencia en estudios de rigidez de flexión de membranas lipídicas con componentes lipídicos individuales 38,72 y múltiples39,66 y en estudios de ablandamiento / rigidez de membranas por inclusiones biológicas (por ejemplo, colesterol, moléculas de medicamentos, péptidos / proteínas)36,37,73,74,75y aditivos sintéticos (por ejemplo, nanopartículas)76,77.
, donde Icoh e Iinc son, respectivamente, la intensidad de dispersión coherente e incoherente de la muestra. Por lo tanto, es aconsejable preparar muestras liposomales de NSE en tampones deuterados (es decir, tampones preparados con D2O en lugar de H2O) para minimizar la señal de dispersión incoherente, principalmente aportada por el contenido de hidrógeno de la muestra. Sin embargo, en algunos casos pueden ser necesarios esquemas intermedios de deuteración (es decir, utilizando mezclas de D2O y H2O) para obtener condiciones óptimas de contraste. Por lo general, las mediciones de NSE de las fluctuaciones de flexión de la membrana se realizan en liposomas totalmente protiated en tampón deuterado, conocidos como liposomas totalmente contrastados en la Figura 5. Este esquema de deuteración da como resultado una gran diferencia de NSLD entre el núcleo de membrana (~0 × 10-6 Å-2) y su entorno de fluido deuterado (~6.4 × 10-6 Å-2), lo que mejora significativamente la señal de dispersión de las membranas liposomales y mejora las estadísticas de medición de la dinámica de flexión. Este esquema decontraste(Figura 2A panel izquierdo) se utiliza con frecuencia en estudios de rigidez de flexión de membranas lipídicas con componentes lipídicos individuales 38,72 y múltiples39,66 y en estudios de ablandamiento / rigidez de membranas por inclusiones biológicas (por ejemplo, colesterol, moléculas de medicamentos, péptidos / proteínas)36,37,73,74,75y aditivos sintéticos (por ejemplo, nanopartículas)76,77.
Las mediciones de las fluctuaciones de flexión dan como resultado tasas de relajación que siguen una dependencia Q3, según lo predicho por Zilman y Granek para láminas delgadas elásticas térmicamente onduladas70. Una forma refinada de esta Q-dependenciase obtiene de las correcciones teóricas de Watson y Brown78,que tienen en cuenta los efectos de la fricción intermonocapa propuestos por Seifert y Langer79. Al definir adicionalmente el plano neutro que se encuentra en la interfaz entre los grupos de cabeza hidrófilos y las colas hidrofóbicas de la membrana, las tasas de relajación de flexión se pueden ajustar a la siguiente expresión38.

donde ηbuff es la viscosidad tampón, kBT es la energía térmica, κ y es la rigidez de flexión de la membrana medida (o de la porción contrastada de la membrana en sistemas deuterados selectivamente). Este tipo de medición permite el cálculo directo de las propiedades elásticas de la membrana en forma del módulo de rigidez de flexión. Nótese que κ se extrae de la pendiente del ajuste lineal de Γ vs. Q3,como se muestra en la Figura 5C.
Por otro lado, las mediciones NSE de las fluctuaciones del espesor de lamembrana muestran desviaciones de la Q3-dependencia en Γ(Q) alrededor de los valores Q que corresponden al espesor de la membrana (ver Figura 2 en ref.66). Para aislar la señal de fluctuación de espesor, se puede dividir Γ(Q)por Q3,como se muestra en la Figura 5D. Los datos resultantes muestran que el exceso de dinámica debido a las fluctuaciones de espesor sigue una función lorentzense en Q,como se corroboró recientemente en simulaciones de dinámica molecular de grano grueso (DM)67. Para ajustarse a la dinámica de exceso observada, Nagao et al.38 desarrollaron una expresión basada en el marco teórico de las fluctuaciones de membrana de Bingham et al.80 de la siguiente manera.

En esta expresión, Q0 es el valor Qmáximo correspondiente al espesor de la membrana (que se puede obtener independientemente de las mediciones de SANS), μ es la viscosidad de la membrana en el plano, AL es el área por lípido (medida con SANS / SAXS) y KA es el módulo de compresibilidad del área. Suponiendo que KA se puede calcular a partir de κ utilizando el modelo de cepillo de polímero, esta expresión se reduce a un parámetro de ajuste, a saber, la viscosidad de la membrana μ,presentando un nuevo enfoque para medir la viscosidad de la membrana sin la necesidad de etiquetado de fluorescencia o anclaje / seguimiento de partículas13. La premisa es que según los modelos de deformación de láminas delgadas elásticas81, κ y KA son interdependientes tales que: ,  donde tm es el espesor mecánico (o deformable) de la membrana y β es una constante que describe el acoplamiento entre hojas. La suposición es que β = 12 para los folletos completamente acoplados, β = 48 para los folletos completamente desacoplados y β = 24 para los folletos acoplados intermediamente. Este último se conoce como el modelo de cepillo de polímero81 y se ha demostrado que se aplica en membranas lipídicas de fluido binario y de un solo componente39. Sin embargo, esto debe abordarse con precaución. Por ejemplo, simulaciones recientes de Doktorova etal. 82 mostró que para que el modelo de cepillo de polímero se mantenga en membranas lipídicas insaturadas que contienen colesterol, se debe utilizar una expresión modificada del espesor de la membrana mecánica. Idealmente, si es posible una medición independiente de KA, por ejemplo, utilizando la aspiración de micropipeta83,entonces la combinación de los resultados de KA con las mediciones de rigidez de flexión NSE presentaría una oportunidad única para investigar el acoplamiento entre perfiles en membranas modelo y biológicas, una pregunta de larga data en biofísica de membranas y biología estructural. Una vez validados los valores de KA, se pueden utilizar en la ecuación 5 para obtener la viscosidad de la membrana mesoscópica.
donde tm es el espesor mecánico (o deformable) de la membrana y β es una constante que describe el acoplamiento entre hojas. La suposición es que β = 12 para los folletos completamente acoplados, β = 48 para los folletos completamente desacoplados y β = 24 para los folletos acoplados intermediamente. Este último se conoce como el modelo de cepillo de polímero81 y se ha demostrado que se aplica en membranas lipídicas de fluido binario y de un solo componente39. Sin embargo, esto debe abordarse con precaución. Por ejemplo, simulaciones recientes de Doktorova etal. 82 mostró que para que el modelo de cepillo de polímero se mantenga en membranas lipídicas insaturadas que contienen colesterol, se debe utilizar una expresión modificada del espesor de la membrana mecánica. Idealmente, si es posible una medición independiente de KA, por ejemplo, utilizando la aspiración de micropipeta83,entonces la combinación de los resultados de KA con las mediciones de rigidez de flexión NSE presentaría una oportunidad única para investigar el acoplamiento entre perfiles en membranas modelo y biológicas, una pregunta de larga data en biofísica de membranas y biología estructural. Una vez validados los valores de KA, se pueden utilizar en la ecuación 5 para obtener la viscosidad de la membrana mesoscópica.

Figura 1: Diseño del instrumento NSE y superposición sinérgica con escalas de longitud/tiempo de la dinámica de membrana mesoscópica. (A) Esquema de los diferentes elementos magnéticos de un instrumento NSE, utilizado para manipular el espín de neutrones que atraviesan el instrumento de izquierda a derecha. El neutrón resaltado indica un cambio en la orientación del espín (o pérdida de polarización) debido al intercambio de energía entre el neutrón y la muestra, mientras que el neutrón transparente representa el espín-eco, es decir, ningún cambio en el espín del neutrón debido al intercambio de energía cero. La flecha gris indica la posibilidad de girar el segundo brazo del espectrómetro para acceder a ángulos de dispersión más grandes. (B) Representación pictórica de la dinámica jerárquica en membranas lipídicas, mostrando varios modos dinámicos que abarcan múltiples escalas de longitud y tiempo. El área sombreada representa las escalas de longitud y tiempo a las que accede NSE, que se superponen con las mesoescalas de las fluctuaciones colectivas de la membrana, a saber, las fluctuaciones de flexión y espesor. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Ejemplos de posibles esquemas de deuteración en experimentos de NSE en membranas lipídicas. (A) Izquierda: Membranas totalmente contrastadas, por ejemplo, membranas protiadas en tampón deuterado, que muestran el perfil NSLD a lo largo de la superficie normal a la membrana. La diferencia en el NSLD entre la región de la cola (~0 × 10-2 Å -2) y la región del grupo de cabeza (~4.5 × 10-6 Å-2) de la membrana se debe a la hidratación del grupo de cabeza con tampón deuterado. Derecha: Membranas emparejadas con contraste de cola de tal manera que la región de cola de hidrocarburo de la membrana tiene el mismo NSLD que el tampón, como se muestra en el perfil NSLD correspondiente a lo largo de la membrana normal. (B) Membranas formadoras de dominios con dos esquemas de contraste de neutrones donde los dominios (centro) o la matriz (izquierda) se emparejan con el búfer, lo que permite estudios selectivos de la dinámica de matrices o dominios, respectivamente. Esta cifra ha sido modificada de Nickels et al., JACS 201541. (C) Membranas asimétricas preparadas por ciclodextrina intercambian entre vesículas lipídicas protiadas y deuteradas, lo que resulta en la deuteración de una valva de membrana mientras se mantiene la otra valva protiated. Esto permite estudiar la dinámica de flexión de la valva protiatada y proporciona información sobre el acoplamiento mecánico entre las valvas opuestas en membranas asimétricas. Esta cifra ha sido modificada a partir de Rickeard et al., Nanoscale 202040. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: Ilustración de la configuración para la extrusión automatizada de liposomas. (A) Extrusora automatizada hecha a medida utilizando una bomba de jeringa, un mini juego de extrusora y un marco de aluminio / acero para permitir extrusiones cíclicas. (B) y (C) muestran la diferencia en la apariencia visual de las suspensiones lipídicas antes (blanco lechoso) y después (azul ópalo transparente) extrusión. Esto se debe a la formación inicial de pilas de lípidos del tamaño de micras o vesículas gigantes que son del orden de, o más grandes que, la longitud de onda de la luz visible. Después de la extrusión, la suspensión comprenderá vesículas nanoscópicas (~ 100 nm), que son más pequeñas que la longitud de onda de la luz visible, produciendo una suspensión transparente. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Datos representativos de experimentos NSE sobre suspensiones liposomales. (A) Ejemplo de una señal de eco sobre un solo píxel detector (píxel marcado en el panel B), que muestra los ajustes de la señal de eco utilizando la ecuación (1), con una ilustración de los diferentes parámetros requeridos en el ajuste de eco. Tenga en cuenta que la señal de eco se traza en función del ángulo de fase en lugar de la corriente de fase como se describe en el paso 4.7 del protocolo. (B) Imagen del detector NSE que muestra la variación en los recuentos de neutrones por píxel. La imagen también muestra píxeles del detector eliminados (verde) debido a las malas señales de eco. El binning de los píxeles del detector en Q-arcs (también conocidos como anillos de Debye-Scherrer) produce la dependencia Q de la función de dispersión intermedia, necesaria para analizar e interpretar datos NSE. Esta cifra fue modificada de Ashkar, J. Appl. Phys. 202050. Haga clic aquí para ver una versión más grande de esta figura.

Figura 5: Resultados representativos de experimentos de NSE en suspensiones liposomales con diferentes esquemas de deuteración. (A) Geometría de dispersión de un neutrón que interactúa con un liposoma, mostrando el ángulo de dispersión, 2θ, y la transferencia del vector de onda,  . (B) Funciones de dispersión intermedias, S(Q,t) / S(Q,0), exhiben desintegraciones en función del tiempo de Fourier. El ajuste de las desintegraciones medidas a una función exponencial estirada dada por la ecuación 3 produce las tasas de relajación, Γ(Q). (C) Para liposomas totalmente protiated en tampón deuterado, Γ(Q) sigue una dependencia Q3, típica de la dinámica de flexión. El ajuste lineal de los datos obtenidos a un modelo de Zilman-Granek produce el módulo de rigidez de flexión de la membrana. (D) Para los liposomas deuterados de la cola, se observa un exceso de dinámica además de las fluctuaciones de flexión y son más pronunciados en valores Q que corresponden al grosor de la membrana. Ajustar el exceso de dinámica a una función lorentziana (ecuación 5) permite la extracción de la viscosidad de la membrana. Los conjuntos de datos se recopilaron en el espectrómetro NSE en el NIST. Haga clic aquí para ver una versión más grande de esta figura.
. (B) Funciones de dispersión intermedias, S(Q,t) / S(Q,0), exhiben desintegraciones en función del tiempo de Fourier. El ajuste de las desintegraciones medidas a una función exponencial estirada dada por la ecuación 3 produce las tasas de relajación, Γ(Q). (C) Para liposomas totalmente protiated en tampón deuterado, Γ(Q) sigue una dependencia Q3, típica de la dinámica de flexión. El ajuste lineal de los datos obtenidos a un modelo de Zilman-Granek produce el módulo de rigidez de flexión de la membrana. (D) Para los liposomas deuterados de la cola, se observa un exceso de dinámica además de las fluctuaciones de flexión y son más pronunciados en valores Q que corresponden al grosor de la membrana. Ajustar el exceso de dinámica a una función lorentziana (ecuación 5) permite la extracción de la viscosidad de la membrana. Los conjuntos de datos se recopilaron en el espectrómetro NSE en el NIST. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
NSE es una técnica poderosa y única en la medición de la dinámica mesoscópica de las membranas lipídicas bajo diversas condiciones. La utilización efectiva de NSE depende de la calidad de la muestra, el contraste de neutrones y el rango de dinámica accesible que se puede sondear para una muestra determinada. Por lo tanto, se requieren varios pasos críticos para realizar experimentos exitosos de NSE y recopilar datos de alta calidad. Un paso clave para garantizar el uso efectivo del tiempo de haz de neutrones durante un experimento de NSE es caracterizar las suspensiones liposomales con métodos basados en laboratorio antes del experimento de NSE. Para exmaple, la distribución de tamaño (o constante de difusión) de los liposomas extruidos puede determinarse mediante dispersión dinámica de la luz (DLS), fácilmente disponible en laboratorios individuales o en instalaciones compartidas84. La microscopía crio-electrónica es otro método de charcaterización recientemente validado en muestras liposomales, donde las imágenes de microscopía de alta resolución en secciones criomicrotomizadas de suspensión liposomal se pueden utilizar eficazmente para examinar la unilamelaridad liposomal65,la formación dedominios 85,86,o la incorporación de aditivos como las nanopartículas76 y las proteínas87. Alternativamente, la dispersión de rayos X de ángulo pequeño (SAXS) se puede utilizar para caracterizar la estructura de la membrana88,evaluar la multilamelaridad liposomal65o evaluar los efectos de los aditivos en las propiedades estructurales de la membrana89. Además de estas técnicas de laboratorio, es muy recomendable que las mediciones de NSE en muestras liposomales se combinen con estudios estructurales utilizando dispersión de neutrones de ángulo pequeño (SANS)54,90. SANS es un excelente complemento de NSE, no solo para adquirir información de membrana estructural, sino también para examinar la intensidad de la señal de dispersión de neutrones de la muestra, confirmar el esquema de contraste y tomar una decisión informada sobre el rango Q sobre el cual se deben realizar las mediciones de NSE. Por lo tanto, se recomienda que los usuarios de NSE soliciten sans beamtime al solicitar experimentos de NSE.
Sin embargo, la NSE sufre de limitaciones muestral en los estudios de membranas biológicas. Uno de los principales factores limitantes de tales experimentos es la cantidad estándar de muestra requerida para las mediciones de NSE (2-4 ml) y las altas concentraciones de muestra que ascienden a 100-200 mg de material de membrana (lípidos y proteínas) para obtener datos de alta calidad. En muchos casos, la producción de tales cantidades de material biológico no es factible o tiene un costo prohibitivo. En tales escenarios, es posible reducir la concentración a 20-25 mg / ml, pero esto requeriría al menos un aumento de 4 veces en el tiempo de adquisición para obtener estadísticas comparables a las muestras con concentraciones de 50 mg / ml. Estos estrictos requisitos sobre el volumen y la concentración de la muestra podrían aliviarse con la próxima generación de espectrómetros NSE en fuentes de neutrones de mayor flujo, como la segunda estación objetivo en el Laboratorio Nacional de Oak Ridge y la Fuente Europea de Espalación. Otra limitación crítica en la realización de experimentos de NSE en membranas lipídicas que requieren esquemas de deuteración selectiva es la falta de disponibilidad comercial de algunas variantes deuteradas de moléculas lipídicas o sus precios exorbitantes, si están disponibles. En algunos casos, estas limitaciones se pueden eludir solicitando la síntesis de lípidos deuterados (o colesterol, proteínas) a través de las instalaciones de deuteración del usuario, como el laboratorio de biodeuteración en el Laboratorio Nacional de Oak Ridge, la instalación nacional de deuteración en ANSTO o la instalación de deuteración en la Fuente de Neutrones y Muones isis. El acceso a estas instalaciones y sus capacidades de síntesis está disponible a través de propuestas de usuarios presentadas que son revisadas por pares sobre la base del mérito científico de la síntesis de material propuesta y su uso previsto en estudios sensibles a los isótopos.
A pesar de estas limitaciones, la aplicación de la espectroscopia NSE en estudios de mecánica de membranas ha llevado a la determinación de los módulos de rigidez de flexión de membranas con diversos grados de complejidad, desde membranas lipídicas monocomponentes35,38 hasta membranas biomiméticas multicomponentes41,66,91, todas las cuales han avanzado nuestra comprensión de la naturaleza dinámica de las membranas lipídicas. Por ejemplo, las mediciones de rigidez de flexión NSE de membranas lipídicas con diferentes unidades moleculares, por ejemplo, lípidos de diferentes longitudes de cadena de acilo y saturación de cadena38,72,92, han proporcionado información esencial sobre el papel de la química molecular en la mecánica de membranas. Cuando se combinan con información estructural, como el espesor de la membrana o el empaquetamiento molecular93,estas mediciones comienzan a proporcionar nuevas perspectivas sobre la interdependencia entre la estructura y la dinámica de la membrana y cómo influyen en la función de la membrana. Las escalas mesoscópicas de NSE lo posicionan de manera única para tales investigaciones fundamentales de relaciones estructura-propiedad, más relevantes en la escala de longitud de los ensamblajes moleculares. Este tema fue explorado recientemente en dos estudios de NSE en membranas lipídicas ricas en colesterol36 y en membranas lipídicas binarias con desajuste hidrofóbico entre los dos componentes lipídicos39. Ambos estudios encontraron pruebas sólidas de que la mecánica de membranas escala con el área por lípido, corroborando las conclusiones de una reciente simulación de MD de todos los átomos por Doktorova et al.82. Estos hallazgos enfatizan la naturaleza autoensamblada de las membranas lipídicas y proporcionan una imagen unificadora del empaquetamiento molecular como un parámetro clave para definir las propiedades dinámicas y funcionales de la membrana.
Otras aplicaciones de la NSE implican estudios de la respuesta mecánica de las membranas a pequeños aditivos, incluidas moléculas biológicas como el colesterol36,37,trehalosa92y melitina73,94,o aditivos inorgánicos como nanopartículas para aplicaciones de administración de fármacos76. NSE también se ha utilizado para comprender cómo la mecánica de membranas responde a los cambios en su entorno, incluida la temperatura92,pH74y la presencia de macromoléculas apiñadas96. Tales estudios están contribuyendo a una mejor comprensión de los factores que influyen en el ablandamiento o endurecimiento de las membranas lipídicas, en condiciones biológicas relacionadas con la salud y la enfermedad, y en entornos controlados para aplicaciones terapéuticas. En particular, las mediciones de NSE también se han utilizado para sondear el efecto de los péptidos antimicrobianos en la dinámica de la membrana73,94,95. Otros ejemplos de aplicaciones de NSE en biomembranas incluyen estudios de la dinámica de estructuras de membrana aplanadas, llamadas tilacoides, que albergan la maquinaria fotosintética en células cianobacterianas97,98.
También se puede utilizar la deuteración selectiva de lípidos en estudios de NSE para investigar la dinámica de características específicas de la membrana que son relevantes para la función biológica. Por ejemplo, Nickels et al. utilizaron la deuteración selectiva de lípidos en membranas lipídicas formadoras de dominios para generar contraste lateral dentro de la membrana, como lo ilustraron previamente Heberle et al.28. Este esquema de deuteración permitió mediciones independientes de la rigidez de flexión de los dominios lipídicos y la matriz lipídica del huésped41 (ver Figura 2B). Los hallazgos confirmaron que los dos compartimentos de membrana tienen módulos de rigidez de flexión distintos, lo que podría ser un mecanismo impulsor para la formación de dominios en las membranas celulares. En un estudio más reciente, Rickeard et al. utilizaron el intercambio de ciclodextrina entre liposomas protiados y deuterados para obtener liposomas asimétricos con folletos etiquetados isotópicamente40 (Figura 2C). Sus liposomas finales tenían una valva protiatada y una valva deuterada que contrasta con el tampón, lo que permite estudios de la dinámica de la valva individual y proporciona una primera explicación experimental directa del efecto de la asimetría y el acoplamiento de la valva en las fluctuaciones de flexión de la membrana.
La deuteración selectiva de membrana también se ha utilizado en estudios de NSE de fluctuaciones de espesor de membrana, un modo dinámico predicho durante mucho tiempo en membranas lipídicas99 que se observó recientemente con el advenimiento de la espectroscopiaNSE 35,100. Estas mediciones utilizan membranas deuteradas de cola para amplificar la señal de las regiones del grupo de cabeza de la membrana y resolver la señal de fluctuación de espesor. Este tipo de experimentos de NSE es relativamente reciente, pero se ha utilizado eficazmente para comprender la interdependencia de las propiedades elásticas y viscosas de la membrana38,para explorar la escala de la rigidez de flexión y la viscosidad con el empaquetamiento molecular en membranas lipídicas mixtas39,y para sondear los efectos locales del colesterol en la viscosidad de la membrana36. Otra área de importancia biológica en la que este modo dinámico podría tener implicaciones de largo alcance son las interacciones mesoscópicas membrana-proteína95. Se sabe que la función de las proteínas de membrana está estrechamente vinculada a la correspondencia hidrofóbica entre la proteína y la membrana del huésped. Por lo tanto, las variaciones en el grosor de la membrana, debido a las fluctuaciones de espesor, podrían actuar como un mecanismo regulador para la función de las proteínas de membrana. NSE es extremadamente adecuado para tales estudios, ya que puede sondear directamente los efectos de la unión e inserción de proteínas en las fluctuaciones del grosor de la membrana. Las mediciones recientes de NSE de nuestro grupo (no publicadas) sugieren que la inserción de proteínas transmembrana podría suprimir significativamente las fluctuaciones del grosor de la membrana y podría presentar un mecanismo potencial para regular los eventos de señalización. Esta es un área de investigación apremiante, pero subdesarrollada, donde la NSE puede tener un impacto significativo en la comprensión de las respuestas dinámicas de las membranas a la unión e inserción de proteínas en las escalas de tiempo y duración de las funciones biológicas clave impartidas por las interacciones de las proteínas con las membranas celulares.
En resumen, la NSE ha evolucionado en los últimos años como una poderosa herramienta para interrogar la dinámica de membranas a escalas espaciales y temporales de funciones biológicas vitales. La técnica está ganando rápidamente un interés generalizado y su potencial para responder preguntas clave en la función de la membrana se está volviendo bien reconocido. Las capacidades de variación de contraste dentro de NSE lo han posicionado como un enfoque único para medir las propiedades de la membrana mesoscópica que de otro modo sería difícil de obtener. Otra ventaja significativa de NSE sobre los métodos tradicionales de espectroscopia en estudios de dinámica de membranas es su superposición con las escalas de longitud y tiempo accesibles con las simulaciones de MD, lo que permite estudios experimentales / de computación sinérgicos para obtener una comprensión a nivel molecular de los diferentes componentes moleculares que componen las membranas. A pesar de su promesa, todavía hay algunas limitaciones en el uso de NSE en estudios de membranas biológicas, incluido el requisito de grandes volúmenes de muestra, la dificultad en la deuteración selectiva en sistemas biológicos y el flujo de neutrones relativamente bajo en los espectrómetros de NSE, lo que resulta en tiempos de medición más largos y disponibilidad limitada de tiempo de haz. Sin embargo, estas deficiencias podrían superarse en un futuro próximo con desarrollos constantes en las fuentes de neutrones y la instrumentación, junto con los avances en las instalaciones de deuteración.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores declaran que no hay conflictos de intereses y no tienen nada que revelar.
Acknowledgments
R. Ashkar gracias M. Nagao, L.-R. Stingaciu y P. Zolnierczuk por muchas discusiones útiles y por su frecuente asistencia con experimentos de NSE en sus respectivas líneas de haz. Los autores reconocen el uso de espectrómetros de eco de espín de neutrones en NIST y ORNL. El espectrómetro NSE en el NIST cuenta con el apoyo del Centro de Dispersión de Neutrones de Alta Resolución, una asociación entre el Instituto Nacional de Estándares y Tecnología y la Fundación Nacional de Ciencias bajo el acuerdo no. DMR-1508249. El espectrómetro NSE en la fuente de neutrones de espalación de ORNL cuenta con el apoyo de la División de Instalaciones de Usuarios Científicos, Oficina de Ciencias Básicas de la Energía, Departamento de Energía de los Estados Unidos. El Laboratorio Nacional de Oak Ridge es administrado por UT-Battelle, LLC bajo el Contrato No. DE-AC05-00OR22725.
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform (biotech grade) | Sigma Aldrich | 496189 | Biotech. grade, ≥99.8%, contains 0.5-1.0% ethanol as stabilizer |
| Circulating water bath | Julabo | SE-12 | Heating Circulator with smart pump, programmable temperature settings, and external sensor connection for measurement and control |
| Deuterium Oxide | Cambridge Isotopes Laboratories | DLM-4 | Deuterated water; Heavy water (D2O) (D, 99.9%) |
| Digital Semi-Microbalance | Mettler Toledo | MS105 | Semi-micro balance with 120 g capacity, 0.01 mg readability, high resolution weighing cell, ergonomic doors, and pipette-check application |
| Ethanol (molecular biology grade) | Sigma Aldrich | E7023 | 200 proof ethanol for molecular biology applications |
| Glass Pipets | VWR | 36360-536 | Disposable Soda Lime glass Pasteur pipets |
| Glass Vials | Thermo Scientific | B7990-1 | Borosilicate glass vials with PTFE/Silione septum caps |
| Lab grade freezer | Fisher Scientific | IU2886D | Ultra-low temprature freezer (-86 to -50 C) for long-term storage of lipids and proteins |
| Lipids (protaited or perdeuterated) | Avanti Polar Lipids | varies by lipid | Lipids can be purchased from Avanti in powder form or in a chloroform solution with the required amounts and deuteration schemes. |
| Millipore water purifier | Millipore Sigma | ZRQSVP3US | Direct-Q® 3 UV Water Purification System which deliver both pure and ultrapure water with a built-in UV lamp to reduce the levels of organics for biological applications |
| Mini Extruder Set | Avanti Polar Lipids | 610020 | Mini-extruder set includes mini-extruder, heating block, 2 GasTight Syringes, and 2 O-rings, Polycarbonate Membranes, and Filter Supports |
| Quick Connect Fittings | Grainger | 2YDA1 and 2YDA7 | Push-button tube fittings for QuickConnect water circulation applications, e.g. high temperature vesicle extrusion |
| Syringe Pump | SyringePump.com | New Era-1000 | Fully programmable syringe pump for infusion and withdrawal; programs up to 41 pumping phases with adjustable pumping rates, dispensed volumes, and extrusion cycles |
| Ultrasonic bath | Fisher Scientific | CPX2800 | Temperature controlled ultra sonic bath with programmable functionality for degassing and ultrasonic applications |
| Vacuum Oven | Thermo Scientific | 3608 | 0.7 cu ft vaccum oven with built-in-high-limit thermostat guards against overheating |
| Vortex Mixer | Fisher Scientific | 02-215-414 | Variable speed, analog control that allows low rpm start-up for gentle shaking or high-speed mixing for vigorous vortexing of samples |
References
- Singer, S. J., Nicolson, G. L. The fluid mosaic model of the structure of cell membranes. Science. 175 (4023), 720-731 (1972).
- Andersen, O. S., Koeppe, R. E. Bilayer thickness and membrane protein function: an energetic perspective. Annual Review of Biophysics and Biomolecular Structure. 36, 107-130 (2007).
- Lundbæk, J. A., Collingwood, S. A., Ingólfsson, H. I., Kapoor, R., Andersen, O. S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. Journal of The Royal Society Interface. 7 (44), 373-395 (2010).
- Bradley, R. P., Radhakrishnan, R. Curvature-undulation coupling as a basis for curvature sensing and generation in bilayer membranes. Proceedings of the National Academy of Sciences of the United States of America. 113 (35), 117-124 (2016).
- Perozo, E., Cortes, D. M., Sompornpisut, P., Kloda, A., Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418 (6901), 942-948 (2002).
- Jensen, M. Ø, Mouritsen, O. G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1666 (1-2), 205-226 (2004).
- Rajendran, L., Simons, K. Lipid rafts and membrane dynamics. Journal of Cell Science. 118 (6), 1099-1102 (2005).
- Katchalsky, A., Spangler, R. Dynamics of membrane processes. Quarterly Reviews of Biophysics. 1 (2), 127-175 (1968).
- Rheinstädter, M. C. Collective molecular dynamics in proteins and membranes (Review). Biointerphases. 3 (2), 83-90 (2008).
- Fujiwara, T., Ritchie, K., Murakoshi, H., Jacobson, K., Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. The Journal of Cell Biology. 157 (6), 1071-1082 (2002).
- Hac, A. E., Seeger, H. M., Fidorra, M., Heimburg, T. Diffusion in two-component lipid membranes--a fluorescence correlation spectroscopy and monte carlo simulation study. Biophysical Journal. 88 (1), 317-333 (2005).
- Heinrich, M., Tian, A., Esposito, C., Baumgart, T. Dynamic sorting of lipids and proteins in membrane tubes with a moving phase boundary. Proceedings of the National Academy of Sciences of the United States of America. 107 (16), 7208-7213 (2010).
- Hormel, T. T., Kurihara, S. Q., Brennan, M. K., Wozniak, M. C., Parthasarathy, R. Measuring lipid membrane viscosity using rotational and translational probe diffusion. Physical Review Letters. 112 (18), 188101 (2014).
- Dimova, R. Recent developments in the field of bending rigidity measurements on membranes. Advances in Colloid and Interface Science. 208, 225-234 (2014).
- Bassereau, P., Sorre, B., Lévy, A. Bending lipid membranes: Experiments after W. Helfrich's model. Advances in Colloid and Interface Science. 208, 47-57 (2014).
- Monzel, C., Sengupta, K. Measuring shape fluctuations in biological membranes. Journal of Physics D: Applied Physics. 49 (24), 243002 (2016).
- Deserno, M. Mesoscopic membrane physics: concepts, simulations, and selected applications. Macromolecular Rapid Communications. 30 (9-10), 752-771 (2009).
- Reynwar, B. J., et al. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 447 (7143), 461-464 (2007).
- Haswell, E. S., Phillips, R., Rees, D. C. Mechanosensitive channels: what can they do and how do they do it. Structure. 19 (10), 1356-1369 (2011).
- Phillips, R., Ursell, T., Wiggins, P., Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature. 459 (7245), 379-385 (2009).
- Dill, K. A., Chan, H. S. From Levinthal to pathways to funnels. Nature Structural Biology. 4 (1), 10-19 (1997).
- Henzler-Wildman, K., Kern, D. Dynamic personalities of proteins. Nature. 450 (7172), 964-972 (2007).
- Grimaldo, M., Roosen-Runge, F., Zhang, F., Schreiber, F., Seydel, T. Dynamics of proteins in solution. Quarterly Reviews of Biophysics. 52, 7 (2019).
- Lyman, E., Hsieh, C. -L., Eggeling, C. From dynamics to membrane organization: experimental breakthroughs occasion a "modeling manifesto". Biophysical Journal. 115 (4), 595-604 (2018).
- Arriaga, L. R., et al. Dissipative curvature fluctuations in bilayer vesicles: Coexistence of pure-bending and hybrid curvature-compression modes. The European Physical Journal. E, Soft Matter. 31 (1), 105-113 (2010).
- Honerkamp-Smith, A. R., Veatch, S. L., Keller, S. L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1788 (1), 53-63 (2009).
- Veatch, S. L., Keller, S. L. Organization in lipid membranes containing cholesterol. Physical Review Letters. 89 (26), 268101 (2002).
- Heberle, F. A., et al. Bilayer thickness mismatch controls domain size in model membranes. Journal of the American Chemical Society. 135 (18), 6853-6859 (2013).
- Nickels, J. D., et al. The in vivo structure of biological membranes and evidence for lipid domains. PLOS Biology. 15 (5), 2002214 (2017).
- Simons, K., Ikonen, E. Functional rafts in cell membranes. Nature. 387 (6633), 569-572 (1997).
- van Meer, G., Voelker, D. R., Feigenson, G. W. Membrane lipids: where they are and how they behave. Nature Reviews. Molecular Cell Biology. 9 (2), 112-124 (2008).
- Liu, S. -L., et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nature Chemical Biology. 13, 268 (2016).
- Rothman, J., Lenard, J. Membrane asymmetry. Science. 195 (4280), 743-753 (1977).
- Ashkar, R., et al. Neutron scattering in the biological sciences: progress and prospects. Acta Crystallographica Section D. 74 (12), 1129-1168 (2018).
- Woodka, A. C., Butler, P. D., Porcar, L., Farago, B., Nagao, M. Lipid bilayers and membrane dynamics: insight into thickness fluctuations. Physical Review Letters. 109 (5), 058102 (2012).
- Chakraborty, S., et al. How cholesterol stiffens unsaturated lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (36), 21896-21905 (2020).
- Arriaga, L. R., et al. Stiffening effect of cholesterol on disordered lipid phases: a combined neutron spin echo + dynamic light scattering analysis of the bending elasticity of large unilamellar vesicles. Biophysical Journal. 96 (9), 3629-3637 (2009).
- Nagao, M., Kelley, E. G., Ashkar, R., Bradbury, R., Butler, P. D. Probing elastic and viscous properties of phospholipid bilayers using neutron spin echo spectroscopy. The Journal of Physical Chemistry Letters. 8 (19), 4679-4684 (2017).
- Kelley, E. G., Butler, P. D., Ashkar, R., Bradbury, R., Nagao, M. Scaling relationships for the elastic moduli and viscosity of mixed lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (38), 23365-23373 (2020).
- Rickeard, B. W., et al. Transverse lipid organization dictates bending fluctuations in model plasma membranes. Nanoscale. 12 (3), 1438-1447 (2020).
- Nickels, J. D., et al. Mechanical properties of nanoscopic lipid domains. Journal of the American Chemical Society. 137 (50), 15772-15780 (2015).
- Mezei, F. Neutron spin echo: A new concept in polarized thermal neutron techniques. Zeitschrift für Physik A Hadrons and Nuclei. 255 (2), 146-160 (1972).
- Hayter, J. B., Penfold, J. Neutron spin-echo integral transform spectroscopy. Zeitschrift für Physik B Condensed Matter. 35 (2), 199-205 (1979).
- Monkenbusch, M., Richter, D. Neutrons in Soft Matter. Imae, T., Kanaya, T., Furusaka, M., Torikai, N. , Wiley. ch6 147-182 (2011).
- Pynn, R. Neutron Spin Echo. Mezei, F., Pappas, C., Gutberlet, T. , Springer. Berlin Heidelberg. 159-177 (2003).
- Holderer, O., et al. The JCNS neutron spin-echo spectrometer J-NSE at the FRM II. Measurement Science and Technology. 19 (3), 034022 (2008).
- Schleger, P., et al. The long-wavelength neutron spin-echo spectrometer IN15 at the Institut Laue-Langevin. Physica B: Condensed Matter. 241-243, 164-165 (1997).
- Holderer, O., Zolnierczuk, P., Pasini, S., Stingaciu, L., Monkenbusch, M. A better view through new glasses: Developments at the Jülich neutron spin echo spectrometers. Physica B: Condensed Matter. 562, 9-12 (2019).
- Farago, B., et al. The IN15 upgrade. Neutron News. 26 (3), 15-17 (2015).
- Ashkar, R. Selective dynamics in polymeric materials: Insights from quasi-elastic neutron scattering spectroscopy. Journal of Applied Physics. 127 (15), 151101 (2020).
- Pasini, S., Holderer, O., Kozielewski, T., Richter, D., Phoenix Monkenbusch, M. J-NSE- Phoenix, a neutron spin-echo spectrometer with optimized superconducting precession coils at the MLZ in Garching. Review of Scientific Instruments. 90 (4), 043107 (2019).
- Svergun, D. I., Koch, M. H. J., Timmins, P. A., May, R. P. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules. , Oxford University Press. (2013).
- Eicher, B., et al. Joint small-angle X-ray and neutron scattering data analysis of asymmetric lipid vesicles. Journal of Applied Crystallography. 50 (2), 419-429 (2017).
- Heberle, F. A., et al. Model-based approaches for the determination of lipid bilayer structure from small-angle neutron and X-ray scattering data. European Biophysics Journal. 41 (10), 875-890 (2012).
- Jaksch, S., Koutsioubas, A., Mattauch, S., Holderer, O., Frielinghaus, H. Long-range excitations in phospholipid membranes. Chemistry and Physics of Lipids. 225, 104788 (2019).
- Jaksch, S., et al. Influence of ibuprofen on phospholipid membranes. Physical Review E. 91 (2), 022716 (2015).
- Armstrong, C. L., et al. Effect of cholesterol on the lateral nanoscale dynamics of fluid membranes. European Biophysics Journal. 41 (10), 901-913 (2012).
- Rheinstädter, M. C., Häußler, W., Salditt, T. Dispersion relation of lipid membrane shape fluctuations by neutron spin-echo spectrometry. Physical Review Letters. 97 (4), 048103 (2006).
- Armstrong, C. L., Häußler, W., Seydel, T., Katsaras, J., Rheinstädter, M. C. Nanosecond lipid dynamics in membranes containing cholesterol. Soft Matter. 10 (15), 2600-2611 (2014).
- Nickels, J. D., et al. Lipid rafts: buffers of cell membrane physical properties. The Journal of Physical Chemistry B. 123 (9), 2050-2056 (2019).
- Michonova-Alexova, E. I., Sugár, I. P. Component and state separation in DMPC/DSPC lipid bilayers: a Monte Carlo simulation study. Biophysical Journal. 83 (4), 1820-1833 (2002).
- Sugár, I. P., Thompson, T. E., Biltonen, R. L. Monte Carlo simulation of two-component bilayers: DMPC/DSPC mixtures. Biophysical Journal. 76 (4), 2099-2110 (1999).
- Mabrey, S., Sturtevant, J. M. Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proceedings of the National Academy of Sciences. 73 (11), 3862-3866 (1976).
- Neutron activation and scattering calculator. , Available from: https://www.ncnr.nist.gov/resources/activation/ (2021).
- Scott, H. L., et al. On the mechanism of bilayer separation by extrusion, or why your LUVs are not really unilamellar. Biophysical Journal. 117 (8), 1381-1386 (2019).
- Ashkar, R., et al. Tuning membrane thickness fluctuations in model lipid bilayers. Biophysical Journal. 109 (1), 106-112 (2015).
- Carrillo, J. -M. Y., Katsaras, J., Sumpter, B. G., Ashkar, R. A computational approach for modeling neutron scattering data from lipid bilayers. Journal of Chemical Theory and Computation. 13 (2), 916-925 (2017).
- Azuah, R. T. DAVE: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. Journal of Research of the National Institute of Standards and Technology. 114 (6), 341-358 (2009).
- Van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Physical Review. 95 (1), 249-262 (1954).
- Zilman, A. G., Granek, R. Undulations and dynamic structure factor of membranes. Physical Review Letters. 77 (23), 4788-4791 (1996).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in model biological membranes measured by neutron spin echo spectroscopy. , Walter de Gruyter, Inc. 131-176 (2019).
- Zheng, Y., Michihiro, N., Dobrin, P. B. Bending elasticity of saturated and monounsaturated phospholipid membranes studied by the neutron spin echo technique. Journal of Physics: Condensed Matter. 21 (15), 155104 (2009).
- Sharma, V. K., Qian, S. Effect of an antimicrobial peptide on lateral segregation of lipids: a structure and dynamics study by neutron scattering. Langmuir. 35 (11), 4152-4160 (2019).
- Boggara, M. B., Faraone, A., Krishnamoorti, R. Effect of pH and Ibuprofen on the Phospholipid Bilayer Bending Modulus. The Journal of Physical Chemistry B. 114 (24), 8061-8066 (2010).
- Lee, J. -H., et al. Thermal fluctuation and elasticity of lipid vesicles interacting with pore-forming peptides. Physical Review Letters. 105 (3), 038101 (2010).
- Chakraborty, S., Abbasi, A., Bothun, G. D., Nagao, M., Kitchens, C. L. Phospholipid bilayer softening due to hydrophobic gold nanoparticle inclusions. Langmuir. 34 (44), 13416-13425 (2018).
- Hoffmann, I., et al. Softening of phospholipid membranes by the adhesion of silica nanoparticles - as seen by neutron spin-echo (NSE). Nanoscale. 6 (12), 6945-6952 (2014).
- Watson, M. C., Brown, F. L. H. Interpreting membrane scattering experiments at the mesoscale: the contribution of dissipation within the bilayer. Biophysical Journal. 98 (6), 9-11 (2010).
- Seifert, U., Langer, S. A. Viscous modes of fluid bilayer membranes. Europhysics Letters (EPL). 23 (1), 71-76 (1993).
- Bingham, R. J., Smye, S. W., Olmsted, P. D. Dynamics of an asymmetric bilayer lipid membrane in a viscous solvent. EPL (Europhysics Letters). 111 (1), 18004 (2015).
- Rawicz, W., Olbrich, K. C., McIntosh, T., Needham, D., Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophysical Journal. 79 (1), 328-339 (2000).
- Doktorova, M., LeVine, M. V., Khelashvili, G., Weinstein, H. A new computational method for membrane compressibility: bilayer mechanical thickness revisited. Biophysical Journal. 116 (3), 487-502 (2019).
- Evans, E., Needham, D. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion and colloidal interactions. The Journal of Physical Chemistry. 91 (16), 4219-4228 (1987).
- Lesieur, S., Grabielle-Madelmont, C., Paternostre, M. T., Ollivon, M. Size analysis and stability study of lipid vesicles by high-performance gel exclusion chromatography, turbidity, and dynamic light scattering. Analytical Biochemistry. 192 (2), 334-343 (1991).
- Heberle, F. A., et al. Direct label-free imaging of nanodomains in biomimetic and biological membranes by cryogenic electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19943-19952 (2020).
- Cornell, C. E., Mileant, A., Thakkar, N., Lee, K. K., Keller, S. L. Direct imaging of liquid domains in membranes by cryo-electron tomography. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19713-19719 (2020).
- Yao, X., Fan, X., Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proceedings of the National Academy of Sciences of the United States of America. 117 (31), 18497-18503 (2020).
- Kučerka, N., Nieh, M. -P., Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1808 (11), 2761-2771 (2011).
- Nielsen, J. E., Bjørnestad, V. A., Lund, R. Resolving the structural interactions between antimicrobial peptides and lipid membranes using small-angle scattering methods: the case of indolicidin. Soft Matter. 14 (43), 8750-8763 (2018).
- Kučerka, N., et al. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophysical Journal. 95 (5), 2356-2367 (2008).
- Kelley, E. G., Butler, P. D., Nagao, M. Scaling of lipid membrane rigidity with domain area fraction. Soft Matter. 15 (13), 2762-2767 (2019).
- Brüning, B. -A., et al. Bilayer undulation dynamics in unilamellar phospholipid vesicles: Effect of temperature, cholesterol and trehalose. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1838 (10), 2412-2419 (2014).
- Kučerka, N., et al. Areas of monounsaturated diacylphosphatidylcholines. Biophysical Journal. 97 (7), 1926-1932 (2009).
- Sharma, V. K., Mamontov, E., Anunciado, D. B., O'Neill, H., Urban, V. S. Effect of antimicrobial peptide on the dynamics of phosphocholine membrane: role of cholesterol and physical state of bilayer. Soft Matter. 11 (34), 6755-6767 (2015).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in lipid membranes containing transmembrane peptides. Soft Matter. , (2021).
- Yu, J., et al. Structure and dynamics of lipid membranes interacting with antivirulence end-phosphorylated polyethylene glycol block copolymers. Soft Matter. 16 (4), 983-989 (2020).
- Stingaciu, L. -R., et al. Revealing the dynamics of thylakoid membranes in living cyanobacterial cells. Scientific Reports. 6 (1), 19627 (2016).
- Stingaciu, L. -R., O'Neill, H. M., Liberton, M., Pakrasi, H. B., Urban, V. S. Influence of chemically disrupted photosynthesis on cyanobacterial thylakoid dynamics in synechocystis sp. PCC 6803. Scientific Reports. 9 (1), 5711 (2019).
- Miller, I. R. Energetics of fluctuation in lipid bilayer thickness. Biophysical Journal. 45 (3), 643-644 (1984).
- Nagao, M. Observation of local thickness fluctuations in surfactant membranes using neutron spin echo. Physical Review E. 80 (3), 031606 (2009).
Tags
Biología Número 171 dinámica lipídica colectiva módulo de flexión compresibilidad del área fluctuaciones de espesor viscosidad de la membrana deuteración selectiva preparación de liposomasErratum
Formal Correction: Erratum: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions
Posted by JoVE Editors on 08/06/2021.
Citeable Link.
An erratum was issued for: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions. The Introduction, Protocol, and Representative Results sections have been updated.
In the Introduction, the fith pargraph was updated from:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 2 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
to:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 0 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
In the Protocol, step 1.1 was updated from:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 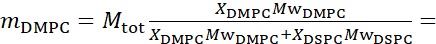 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
to:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 0 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
In the Representative Results, the fist pagargaph was updted from:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: 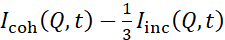 , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
, where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
to:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~0 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
In the Representative Reults, Figure 2 was updated from:
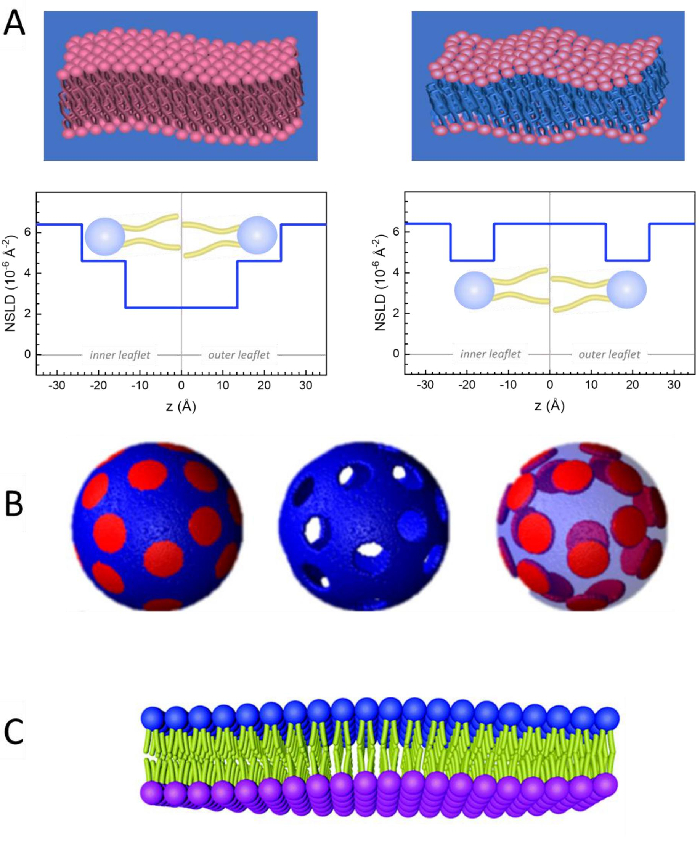
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the headgroup (~2 × 10-2 Å-2) and tail region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
to:
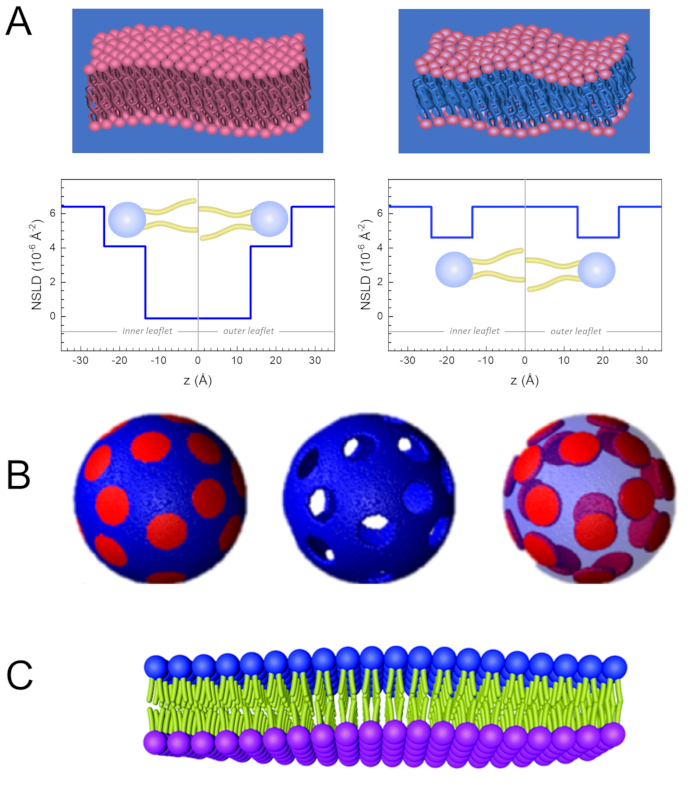
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the tail region (~0 × 10-2 Å-2) and headgroup region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
