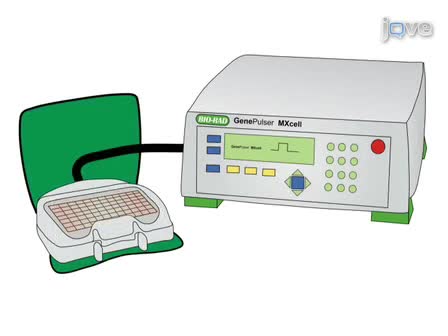
Genetic associations often remain unexplained at a functional level. This method aims to assess the effect of phenotype-associated genetic markers on gene expression by analyzing cells heterozygous for transcribed SNPs. The technology allows accurate measurement by MALDI-TOF mass spectrometry to quantify allele-specific primer extension products.