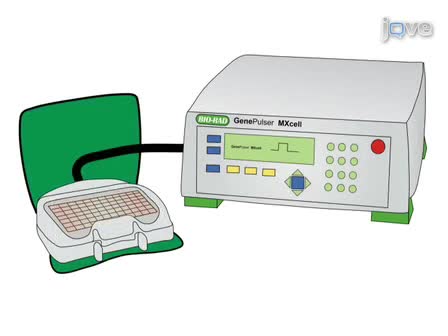
This article describes the procedure for the identification and characterization of a gene family in grapevine applied to the family of Arabidopsis Tóxicos in Levadura (ATL) E3 ubiquitin ligases.