

Summary
神话允许的瞬时和稳定的互动模式生物酵母中表达的蛋白质之间的灵敏的检测。它已成功地应用于研究外源性和酵母的完整的膜蛋白,以确定其在高通量的方式相互作用的合作伙伴。
Abstract
基本的生物和临床不可或缺的膜蛋白的重要性,提示为全长跨膜蛋白的蛋白质相互作用(PPI)的高通量鉴定酵母为基础的系统的发展。为此,我们的实验室开发的分裂泛基于膜酵母双杂交系统(神话)。这种技术允许的瞬时和稳定的蛋白质相互作用的敏感检测
Protocol
1。背景资料
蛋白质 - 蛋白质相互作用(生产者价格指数)是参与所有细胞过程的基本构建块。因此,它是必不可少的,所有交互严格管制,以维持细胞的动态平衡,在这个生物平衡的转变,通常扮演在疾病和癌症细胞转化的作用。膜相关蛋白是生物最重要的一类蛋白质之中,因为他们可以启动复杂的信号级联,并进行调解的进口和出口的各种分子,包括药物,被列为相关的问题,最近在卫生保健领域的意义耐药性已成为越来越普遍。获得洞察到这种蛋白质类的复杂性,需要确定其交互伙伴。发现这样的合作伙伴已被证明具有挑战性,因为它往往需要苛刻的条件,必须为每个膜结合蛋白进行了优化[1]。
基本的生物和临床不可或缺的膜蛋白的重要性促使了PPI的高通量鉴定全长跨膜蛋白的酵母为基础的系统的发展。为此,我们开发了基于分裂泛素膜酵母双杂交系统(神话)[2-4]。这个工具允许瞬时和稳定的蛋白质相互作用的敏感检测。它已成功地应用于研究的模式生物酿酒酵母中表达的外源性和内源性蛋白[3-7 ]。神话的泛素可能被分隔成两个基团的观察:C -末端的一半 (UB)和N -末端的一半( 努比)在体内研究表明,这些基团自发重组,由于其高亲和力另一个(Figure. 1A)。然而,引进异亮氨酸13甘氨酸在N -端的泛素一半的点突变(生产简称为N UB摹的一个片段)防止这种自发的重新关联[8](Figure. 1B) 。
我们在神话系统(Figure. 1C和2)使用了这一原则。简言之,积分膜诱饵蛋白融合到C UB基团,这是与人工转录因子的大肠埃希氏菌DNA结合蛋白LexA的组成,由单纯疱疹病毒VP16的激活域。猎物产生融合基因或基因组DNA片段的NubG基团。诱饵和猎物蛋白在酵母宿主之间的相互作用,导致重组一个完整长度的“伪泛素”分子,随后承认胞质deubiquitinating酶(配音)和转录因子的蛋白水解释放。转录因子可以进入细胞核,激活报告基因系统(通常涉及HIS3,lacZ的ADE2基因的表达)允许选择性培养基上生长的酵母菌株,这是表明诱饵和猎物的相互作用[ 2-4]。
整体膜蛋白相互作用的识别和表征将提供信息,帮助客户充分了解它们的功能。正如我们更准确地理解和剖析的互动与整合膜蛋白的蛋白质,的角色,我们可以争取到参与调节这些蛋白质的动态相互作用的洞察力和发现新的目标,可能具有治疗潜力。
2。诱饵和适当的神话系统的选择
- 开展谎言分析之前,验证你的蛋白在细胞的细胞质中的N -和/或C -末端。它融合的蛋白质的C UB - LexA - VP16的标签,在这样一个总站,自deubiquitinating转录因子的释放所需的酶是位于细胞质中,是必不可少的[ 4]。
- 接下来,决定神话的两个主要变种,这是合适的。对于非母语的酵母蛋白,可用于传统的神话(tMYTH),其中诱饵异位过度表达质粒。对于原生酵母蛋白,综合神话(iMYTH)是首选的方法。 iMYTH毒饵内源性标记与C UB - LexA - VP16的标签,他们离开其原生启动子的控制下。这是有利的,因为野生型的诱饵的表达水平,有助于消除与蛋白过度表达,如误报的数量增加[3],有关的问题。初始诱饵的建设和使用的选择性培养基外,神话的两种形式,在本质上是相同的方式进行。为了清楚起见,我们将主要侧重于在本报告中使用的tMYTH,这种变异可以在原则上,可用于膜蛋白从几乎任何生物体是从而更广泛适用。
3。诱饵的生成和验证
- 所需的媒体和解决方案
- 准备在121℃,15磅30分钟高压灭菌的无菌DDH 2 。
- 3 -氨基- 1,2,4 -三唑(3 - AT)解决方案作为一个1M的股票解决方案准备在 DDH 2 O。通过消毒,通过0.2微米的过滤器。
- YPAD生长介质组成的W / V 1%酵母提取物,2%W / V蛋白胨,2%W / V葡萄糖和100微米腺嘌呤准备在DDH 2 O在121℃,15磅30分钟高压灭菌消毒。
- 10氨基酸/碱基组合:完整的组合包含1.0毫米1.0毫米腺嘌呤,尿嘧啶1.8毫米,1.0毫米精氨酸,组氨酸,异亮氨酸2.3毫米,7.6毫米亮氨酸,赖氨酸1.6毫米,10.1毫米蛋氨酸,苯丙氨酸3.0毫米,16.8毫米苏氨酸,色氨酸2.0毫米,1.7毫米的酪氨酸和12.8毫米缬氨酸,准备在DDH 2 O 。为辍学省略了必要的氨基酸(S)和/或碱基(S)。在121℃,15磅30分钟高压灭菌消毒。
- 合成辍学(SD)的 0.67%W / V酵母氮基(不含氨基酸,但硫酸铵),2%W / V葡萄糖,2%W / V琼脂和1X氨基酸/核苷酸混合组成的生长介质,准备在DDH 2 O。准备液体和固体(含2%琼脂),SD -亮氨酸媒体。还准备坚实的SD -色氨酸,亮氨酸和SD -色氨酸亮氨酸,腺嘌呤,组氨酸媒体。在121℃,15磅30分钟高压灭菌消毒。倒入100x15毫米培养皿固体培养基。
- 合成辍学(SD)的生长介质,含3 - AT。准备SD -色氨酸,亮氨酸,腺嘌呤,组氨酸媒体描述的那样,但含有3 - AT浓度25,50,75和100毫米。添加适量的3 - 1M无菌原液媒体后,已灭菌和冷却(但尚未凝固)。倒入100x15毫米培养皿。
- PEG /醋酸锂混合组成的40%W / V PEG - 3350,120毫米醋酸锂和167微克/毫升鲑鱼精DNA(III型钠盐)准备在DDH 2 O为了确保这种混合的无菌准备无菌水和解决方案(即蒸压50%的PEG - 3350,高压灭菌在无菌 DDH 2准备的1M醋酸锂和2 mg / mL的鲑鱼精子DNA的III型钠盐)。
- 酶和试剂进行PCR扩增 。
- 商业小量制备试剂盒。
- 钠钙玻璃珠(0.5毫米)。
- 主管大肠杆菌细胞适合质粒的传播 (如DH5α中,XL10金)和细菌传播和质粒选择合适的标准媒体的。
- 特定的酵母菌株 ,在协议中所述的质粒和引物。
- 一代峡维修tMYTH诱饵
- 诱饵必须克隆到一个适当的标记和表达载体。目前可用于各种tMYTH向量使用诱饵建设。如pCMBV4,pAMBV4和pTMBV4载体允许控制下的CYC1(弱),ADH1(强)和TEF1(很强)发起人,分别建设的C端标签的诱饵(诱饵- UB - LexA - VP16) 。可以产生N末端 标记诱饵(LexA - VP16 - UB -诱饵),如pTLB - 1和pBT3 - N,TEF1 CYC1促销员的控制下,分别使用的载体。矢量的选择取决于诱饵,必须凭经验确定。在某些情况下更高的诱饵表达是必要的,以便及时发现相互作用,而在其他情况下诱饵的过度表达可能实际上是有害的,导致误报的数量增加。
- 酶切在选定合适的酶切位点(S)的质粒。卵裂应该只发生在紧邻的C UB - LexA - VP16的标签(上游的C -末端标记或标记为N -末端标记下游)。例如,在使用pAMBV4载体时,SfiI是一个理想的选择。存放在-20 ° C,直到准备使用消化质粒。
- 设计引物扩增感兴趣的基因的克隆。您的正向引物5'端必须匹配的酶切位点上游约35-40个核苷酸,而3'端必须符合第18-20核苷酸的靶基因。反向引物5'端必须匹配的酶切位点下游约35-40个核苷酸的反向互补,3'端匹配反向补充最后的靶基因的18-20核苷酸(省略终止密码子如果在C UB - LexA - VP16标签被放置在C -末端)。取决于是否的N -或C -端子标记正在执行中,选择正向或反向引物等靶基因的克隆与C UB - LexA - VP16的标签帧的35-40个核苷酸。
- 兴趣使用上述引物通过PCR扩增基因。将取决于特定的酶,并用特异性引物PCR技术参数。
- 变换轴承LEU2突变(如BY4741)到一个合适的酵母实验室应变消化质粒的PCR产物。一个神话记者株(如THY.AP4或L40)可以使用,但是没有必要的,因为酵母在这一点上的目的是作为一个环境差距修复的同源重组可以发生。转型应进行如下:
- 您所选择的酵母菌株的一个单菌落接种到5毫升无菌YPAD媒体孵育过夜30 ° C(200转)不断晃动。
- 过夜培养稀释成50毫升新鲜YPAD媒体〜0.15的OD600值和孵育30 °晃动(200转)的彗星。成长,直到〜0.6 OD600值达到大约3-4小时。
- 在700xg离心5分钟,去除上清细胞。
- 悬浮在25毫升无菌DDH 2 O的细胞沉淀700xg离心5分钟。
- 去除上清,重悬细胞沉淀在1 mL无菌DDH 2 O
- 一个离心管中加入100μL的细胞,300μL的聚乙二醇/醋酸锂混合,消化质粒(50 fmol的)和PCR产物(250-500 fmol的)。
- 在30 ° C孵育30分钟。
- 热休克42 ° C时为1小时。
- 在3000xg离心5分钟,去除上清。
- 悬浮在200微升无菌DDH 2的细胞沉淀和板的整个卷到固体,SD -亮氨酸的选择性培养基。在30 ° C,成长为2-4天。
- 长大了5毫升液体介质SD -亮氨酸的转化菌株在30℃过夜的单个菌落。
- 在700xg离心5分钟,去除上清细胞。
- 使用任何商业小量制备试剂盒从细胞颗粒诱饵质粒DNA隔离。按照一个修改的标准协议。为了确保有足够的的酵母细胞裂解液,添加一个0.5毫米钠钙玻璃珠的体积小的颗粒后最初的再悬浮和涡大力为5分钟。然后进行正常的商业协议。
- 转换成一个称职的大肠杆菌分离出酵母DNA 菌株适合质粒传播了至少1 × 10 7细胞/微克DNA的转化效率(例如DH5α中,XL10金) 。注意,可以选择使用卡那霉素诱饵质粒。
- 收获转化大肠杆菌质粒DNA使用一个标准的DNA提取方法或商业套件的大肠杆菌。
- 诱饵质粒,经测序验证正确的施工。
- 变换应变,L40)(如THY.AP4建设成一个适当的神话记者核实诱饵。可以用刚才所描述的酵母转化协议,以诱饵质粒DNA消化质粒和PCR产物的地方。
- 诱饵验证 - 正确的本地化
- 在使用之前,诱饵菌株进行了分析,以确保他们正确地本地化的酵母膜。在使用iMYTH时,这种定位将取决于具体的属性标签的诱饵后。 tMYTH,诱饵质粒一般包括一个信号序列(如Matα)质膜指导表达的蛋白质。本地化是用荧光显微镜。列入一个诱饵标签序列YFP的分子(即C UB - YFP - LexA - VP16的)是最简单,最直接的办法,允许直接观察活细胞,并通常在iMYTH使用。另外,一个标准的使用对LexA或VP16的组件的标记抗体的免疫荧光方法可以使用。
- 诱饵验证- N的UB G / I控制测试
- 一旦适当的诱饵本地化已经成立,它是必要的,以确保诱饵不激活记者系统单独或在非相互作用的猎物(即验证诱饵是不是自我激活)存在。这是使用的N UB G / I测试,诱饵的转化与互动(正)和非交互(负)控制猎物,和评估选择性培养基上生长。诱饵我必须选择性培养基上生长n为阳性对照存在,并没有增长中存在阴性对照,以适合使用在神话。
- 首先转化的诱饵应变测试与控制猎物质粒100-200吴。可以使用前面描述的酵母转化协议,代SD -亮氨酸YPAD地方媒体和使用SD -色氨酸,亮氨酸媒体最终电镀步骤。常用以下控制猎物构造:
- UB我POST1 - N(阳性对照)
- POST1 - N的UB G(阴性对照)
- UB我pFUR4 - N(阳性对照)
- pFUR4 - N的UB G(阴性对照)
- OST1 oligosaccharyl复杂的一个组成部分,是本地化的内质网膜[9]而FUR4是尿嘧啶通透和本地化质膜[10]。虽然这些蛋白质通常是我们作为非交互猎物使用的首选,他们是否适合上按个别的情况下会有所不同。万一你的饵是预测真正的互动与这两个控制,将需要选择替代猎物。召回的N UB我是野生型泛素的N -末端的形式,并自发地与C UB 独立的C UB和N UB融合蛋白之间的相互作用的交互。因此ñ UB我囊中物构成,而N UB摹猎物(账面异亮氨酸13,甘氨酸突变,防止自发的N UB和C UB协会)作为阴性对照为阳性对照。
- 重悬于100μL,无菌DDH 2 O到每个转化诱饵单菌落
- 串行稀释在无菌DDH 2悬浮细胞产生稀释1 / 10,1 / 100和1 / 1000。
- SPOT 5μL未稀释和稀释细胞的浓度范围在3 - AT和无到SD -色氨酸,亮氨酸和SD -色氨酸,亮氨酸,腺嘌呤,组氨酸媒体卷。 3 - AT作为HIS3报告基因的竞争性抑制剂的行为,并有助于增加遴选过程的严格。它可以在某些情况下可用于抑制非特定生长弱到中度自我激活毒饵。
- 允许的斑点,干燥,然后在30℃孵育2-4天的板。
- 全部转化生长在SD -色氨酸,亮氨酸板,表明他们已成功地与猎物质粒转化。诱饵其中不自我激活SD -色氨酸,亮氨酸,腺嘌呤,组氨酸媒体将增长只有当与N UB转化,我的猎物结构,以及不与N UB摹猎物。注意浓度的3 - AT是需要在媒体上(如果有的话),因为这将需要在筛选过程中使用。
4。筛选
- 所需的媒体和解决方案
- 准备在121℃,15磅30分钟高压灭菌的无菌DDH 2 。
- 准备在DDH 2 0.9%NaCl溶液和121高压灭菌消毒℃,15 PSI为30分钟。
- 磷酸钠溶液 493 mM磷酸钠二元和250毫米的磷酸二氢钠组成的DDH 2 O在121℃,15磅30分钟高压灭菌消毒。
- X - Gal的(5 -溴-4 -氯-3 -吲哚基-β- D -半乳糖苷)解决方案作为一个在N,N -二甲基甲酰胺100毫克/毫升原液准备。
- 2xYPAD生长介质中含有2%W / V酵母提取物,4%W / V蛋白胨,4%W / V葡萄糖和100微米腺嘌呤,准备在DDH 2 O在121℃,15磅30分钟高压灭菌消毒。
- 合成差(SD)生长介质,准备如前所述。准备液体SD -亮氨酸和坚实的SD -色氨酸亮氨酸。固体培养基倒入两个100x15毫米培养皿。准备在150毫米的圆形板,16板为每个屏幕含有3 - AT,从N UB G / I对照试验确定的浓度,如果必要的话,固体SD -色氨酸,亮氨酸,腺嘌呤,组氨酸媒体。
- 合成差(SD)媒体+ 5 -溴-4 -氯- 3 - Indoyl -β- D -半乳糖苷(X - Gal的)。准备的SD -色氨酸,亮氨酸,腺嘌呤,组氨酸媒体含有琼脂如前所述。高压灭菌后,允许冷,加3 - AT(如有必要),其次是十分之一体积Øf无菌磷酸钠解决方案。接下来,X - Gal的解决方案添加到终浓度为80微克/毫升。调匀,倒入150毫米的圆形板。
- 聚乙二醇/醋酸锂溶液II含40%PEG - 3350,100毫米醋酸锂,1毫米EDTA和10毫米的Tris pH值7.5。准备这个解决方案使用无菌DDH 2 O和解决方案(如蒸压50%PEG - 3350,1 M醋酸锂,100毫米的Tris pH值7.5和500毫米EDTA pH值8.0)。
- 醋酸锂/ TRIS EDTA溶液含有110毫米醋酸锂,11毫米的Tris pH值7.5和1.1毫米EDTA。准备使用无菌DDH 2 O和解决方案(如蒸压1M醋酸锂,100毫米的Tris pH值7.5和500毫米EDTA pH值8.0),这个解决方案。
- 10X三EDTA溶液组成的100毫米的Tris pH值7.5和10 mM EDTA准备在DDH 2 O在121℃,15磅30分钟高压灭菌消毒。
- 单链载体的DNA(单链DNA)的溶液中含有为2 mg / mL的三文鱼精子DNA III型钠盐,准备在无菌DDH 2 O
- 商业小量制备试剂盒。
- 钠钙玻璃珠(0.5毫米)。
- 主管大肠杆菌细胞适合质粒的传播 (如DH5α中,XL10金)和细菌传播和质粒选择合适的标准媒体的。
- 在协议中所述的具体的酵母菌株和质粒
- 大规模改造
- 单个菌落接种一个包含您的诱饵,到5毫升的SD -亮氨酸媒体记者的神话应变,并在30℃孵育过夜用颤抖的(200转)彗星。
- 过夜培养稀释成200毫升SD -亮氨酸媒体一个OD600值= 0.15,并培育30 °(200转)用颤抖的彗星。成长,直到OD600 = 0.6 - 0.7(约4-5小时)。
- 前不久目标OD600值达到解冻等份的单链DNA溶液。熬在100 ° C下5分钟,然后在冰酷。再重复一次。
- 当OD600目标已经达到,收获在700xg细胞通过离心5分钟(4x50毫升螺旋盖离心管之间的鸿沟200 mL培养)。
- 30 mL无菌DDH 2 O和简要涡样品每个洗涤沉淀。在700xg离心5分钟。
- 弃上清,每1毫升醋酸锂/三EDTA溶液重悬沉淀。转移到无菌的1.5 mL离心管中,并在700xg离心5分钟。
- 弃上清,每600μL醋酸锂/三EDTA溶液重悬沉淀。
- 4x15毫升螺旋盖离心管中加入以下:
- 2.5 mL的聚乙二醇/醋酸锂解决方案II
- 600μL的悬浮细胞
- 100μL的单链DNA溶液
- 7微克猎物库DNA
- 库包含标记的N -或UB摹与N的C总站的猎物,并从cDNA或基因组来源的各种准备, 市售 (www.dualsystems.com )。在逐案基础上,取决于所选的诱饵和实验的目标,必须确定所使用的特定库。
- 1分钟的涡管,以确保充分混合,然后在30℃水浴孵育45分钟。混合简要每隔15分钟。
- 每管加入160μL二甲基亚砜(DMSO),并立即反相管混合。
- 在42℃水浴孵育20分钟。
- 收集转化700xg离心5分钟。
- 上清液。收回再悬浮在3 mL 2xYPAD每个颗粒的转化。池中的所有样品,在一个50毫升的螺丝帽的离心管。
- 在30 ° C孵育90分钟细胞恢复。
- 在700xg离心5分钟,弃上清。
- 细胞沉淀重悬在4.9毫升无菌0.9%氯化钠。
- 使用100μL重悬细胞,准备在无菌0.9%氯化钠10倍连续稀释,从10倍到1000倍。
- 板100μL到SD -色氨酸,亮氨酸媒体和孵化的100倍和1000倍稀释液,在30 ° C时2-3天。这些板块作为一个控制和用于计算转化效率。
- 平均分配到大(150毫米)SD -色氨酸,亮氨酸,腺嘌呤,组氨酸板剩余的4.8毫升悬浮细胞和板,在30 ° C含必要数量的3 - AT在N UB /我测试确定,并培育3-4天。
- 重悬于100μL0.9%氯化钠和5盘上微升等分单菌落(分别代表细胞含有一个潜在的相互作用诱饵捕食对)SD -色氨酸,亮氨酸,腺嘌呤,组氨酸+ X - Gal的媒体(包括3 - AT如果需要)。允许成长为2-4天。这一步作为一个选择性筛选的第二轮,有助于移除在最初一轮获得的误报。选择作进一步的分析显示强劲增长和蓝色的,只有殖民地。
- 猎物的DNA分离和测序
- 从蓝色的酵母使用与前面介绍的修改的一个小量协议的殖民地质粒DNA隔离。请务必成长起来的细胞只保留猎物的选择,但不是诱饵,质粒在SD -色氨酸媒体。对于产生非常大量的命中的屏幕,市售的高通量小量制备试剂盒可在这一点上是有利的。
- 转换成一个称职的大肠杆菌分离出酵母质粒DNA 菌株适合质粒传播了至少1 × 10 7细胞/微克DNA的转化效率(例如DH5α中,XL10金) 。注意,可以选择使用氨苄青霉素猎物质粒。
- 从转化大肠杆菌质粒DNA隔离使用一个标准的DNA提取方法或商业套件的大肠杆菌。再次,一种高通量的小量制备试剂盒可能是有用的,如果样本数量很大。在大肠杆菌中扩增的DNA 大肠杆菌大大增加质粒产量和确保足够数量的DNA测序,并进一步分析。
- 按顺序使用一个序列互补的引物内的N UB G.孤立的质粒
- 整理和分析所有测序数据的组装您的interactors的初步清单。这可能是手工完成,或在一个自动化的方式,使用适当的软件。
- 诱饵依赖测试
- 组装interactors初步名单后,重要的是重新检查的相互作用和消除交互/激活的方式诱饵身份独立记者系统的混杂猎物。这是通过使用诱饵依赖测试。在这项测试中,所有确定interactors转换回成原来的诱饵应变,以及应变窝藏一个跨膜域融合到C UB - LexA - VP16的标签组成的一个控制人工饵料。按照前面所述的标准协议进行改造,使用SD -亮氨酸媒体YPAD和固体介质SD -色氨酸,亮氨酸,最终电镀步骤。
- 重悬于100μL无菌DDH 2 O和现场上述转换的单菌落到SD -色氨酸,亮氨酸,腺嘌呤,组氨酸+ X - Gal的媒体(包括3 - AT,如果需要的话)5μL卷。板,然后培养2-4天,在30 ° C理想的情况下应选择多个转化为每个猎物,无论是原饵料和人工饵料应察觉到同一板块。
- 酵母携带的人工饵料和猎物,引起了记者的系统(即增长和蓝色)的激活被认为是混杂的interactors名单中删除特定的猎物。
- 导致诱饵利益,而不是人工饵料和酵母生长的蓝色着色的猎物,证实了这一点具体的互动。但是,如果酵母窝藏的猎物,你的饵的利益不长,这个猎物interactors列表中删除。
- 剩余的猎物构成的完整列表在神话屏幕确定interactors。
5。进一步的研究
一旦谎言筛选已经完成,必须进一步分析,以验证并确定检测相互作用的生物学意义。要执行的具体的研究会有所不同在逐案基础上,必须由个别研究人员确定。后续工作的一些常见的例子包括:在原生生物体的免疫共沉淀实验和删除的研究。此外,所获得的数据进行计算分析,可用于检测模式,并帮助确定潜在的相关性和不同的相互作用可能发挥的作用。因此,神话技术作为一个强大的第一步“,对识别和膜蛋白的关键功能上的相互作用的理解。再加上详细的后续研究,以及其他近期发达国家和新兴的特chnologies,它的承诺是一个有价值的工具,在开锁的细胞的奥秘。

图1。分裂泛。A.原理泛素可分为两个基团的C -末端的一半(UB)和N -末端的一半(N UB )。这些基团重组为一个自发的,因为他们的高亲和力。 B. UB我在异亮氨酸甘氨酸13位点突变(N UB G)的防止这种自发的重新关联。 C.在神话体系中,C UB利益诱饵(二)融合和融合到N UB G(A)的猎物。 AB蛋白质相互作用的重组伪泛。
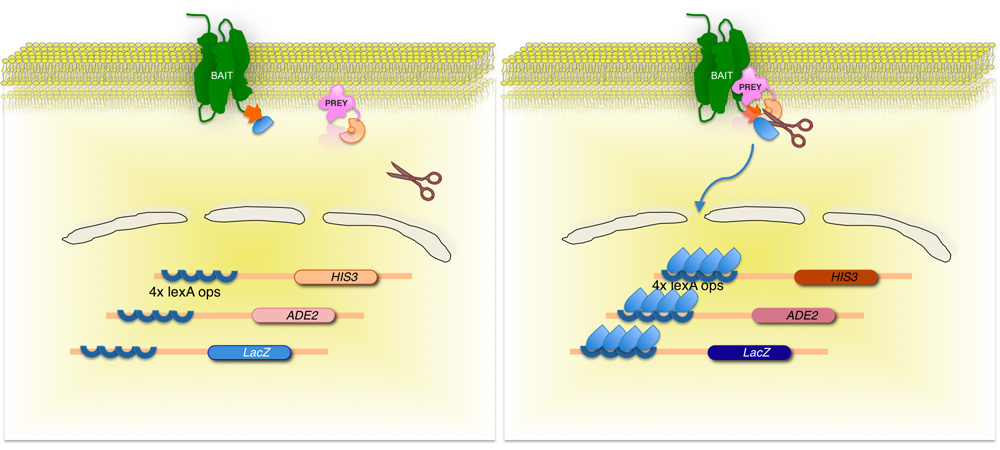
图2。拆分泛基于膜酵母双杂交系统(神话)。
膜蛋白的利益(诱饵)是融合的酵母泛素C -末端的一半(UB),偶联的转录因子。使用cDNA或基因组DNA库,每个库(猎物)编码蛋 白质是相应的N -末端的泛素基团(N泛G)的融合。如果这两种蛋白质不相互作用,转录因子仍然在膜接口(左侧面板)。但是,如果蛋白质相互作用,两个泛基团的加入,导致泛素特异性蛋白酶裂解。卵裂发布的转录因子,从而导致报告基因的表达(右图)

图3。谎言管道。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
神话是第一个高通量确定全长膜蛋白和胞浆或膜结合的合作伙伴之间的的相互作用的系统,使。它已被用于研究膜蛋白的生物体[3-7]。有,但是,具体的细节,可能需要经过严格审核的程序,以确保蛋白质的利益服从研究神话。
许多膜结合蛋白裂解产生的成熟蛋白的信号序列,随后通过质膜。这个序列是生物体的特定的和有可能,这个原始信号序列在酵母中表达时,会导致定位不当利益的蛋白质,因为信号序列仍然无法识别。要绕过这个问题,我们设计了融合酵母交配因子-α(MATα)所得的信号序列,这些特定的蛋白质。这种肽序列(即所谓的MFα- SS)重新定位蛋白酵母质膜和重要的,是由酵母信号肽酶裂解。这种肽序列中发现质粒pTMBVMFαpAMBV -MFα。
需要强调的另一个重要参数是诱饵的表达水平。启动子驱动的诱饵表达调节这个参数。它可能需要优化诱饵的表达水平,使用NubG /努比测试和量必须消除过度表达的文物,诱饵“自我激活”(即胡乱许多非特异性的猎物蛋白质相互作用) 。检查酵母蛋白生产应用iMYTH时的浓度有关生理的诱饵。在这种情况下,标记基因的利益与崽- TF内的基因组DNA。另外,外源蛋白可表示pBT3 STE和pCMBV质粒进行CYC1发起人,造成低诱饵表达。质粒pTMBV pTLB1海港TEF1 pAMBV ADH1启动子的推动者,同时,该驱动器诱饵蛋白的强表达。然而,如果诱饵蛋白水平需要进一步优化,可能有必要使用pTLB - 1质粒进行TEF1发起人,LexA DNA结合结构域突变R156G,以减轻对外生记者基因启动子的亲和力,最终降低自激活的概率[5]。
谎言的成功起着重要作用的另一个因素,是首选的筛选过程中使用的库。这将取决于内源性饵料表达谱。例如,可能会表示诱饵,在特定的组织,因此重要的是要使用一个库,就是从这个特定的组织构造。这将确保有关生理的相互作用被发现。
神话系统是一个简单而快速的工具,提供了丰富的一类已很难研究的蛋白质的信息。这些确定的相互作用,可能有助于澄清的完整膜蛋白的生物学功能。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
伊戈尔Stagljar是DualSystems生物技术,瑞士的创始人之一。
Acknowledgments
我们想感谢黎明埃德蒙兹关键读了这篇稿子。 Stagljar实验室是由从加拿大创新(CFI),加拿大卫生研究所(CIHR),心脏及中风基金会,加拿大癌症协会,和诺华基金会的资金支持。
Materials
| Name | Company | Catalog Number | Comments |
| Polyethenlene Glycol (PEG3350) | BioShop Canada | PEG335 | |
| Lithium Acetate Bihydrate | BioShop Canada | LIA001 | |
| X-Gal (5-Bromo-4-Chloro-3-Indolyl-b-D-galactopyranoside) | BioShop Canada | XGA001 | |
| N`,N-dimethyl formamide | BioShop Canada | DMF 451 | |
| 3-amino-1,2,4-triazole (3-AT) | BioShop Canada | ATT124 | |
| Sodium phosphate dibasic | BioShop Canada | SPD307 | |
| Sodium phosphate monobasic | Fisher Scientific | BP329-500 | |
| Salmon Sperm DNA | VWR international | CA80601-120 | |
| D-Glucose | BioShop Canada | GLU501 | |
| LB Broth LENOX | BioShop Canada | LBL405 | |
| Yeast Nitrogen Base | BioShop Canada | YNB406 | |
| Yeast Extract | BioShop Canada | YEX401 | |
| Peptone | BD Biosciences | 211677 | |
| Bio-Tryptone | BioShop Canada | TRP402 | |
| Adenine Sulphate | BioShop Canada | ADS201 | |
| L-Uracil | BioShop Canada | URA241 | |
| L-Threonine | BioShop Canada | THR002 | |
| L-Histidine | BioShop Canada | HIS200 | |
| L-Methionine | BioShop Canada | MET222 | |
| L-Valine | BioShop Canada | VAL201 | |
| L-Phenylalanine | BioShop Canada | PHA302 | |
| L-Isoleucine | BioShop Canada | ISO910 | |
| L-Tyrosine | BioShop Canada | TYR333 | |
| L-Leucine | BioShop Canada | LEU222 | |
| L-Arginine | BioShop Canada | ARG006 | |
| L-Tryptophane | Fisher Scientific | BP395-100 | |
| L-Lysine | BioShop Canada | LYS101 | |
| L-Alanine | Fisher Scientific | BP369-100 | |
| Agar | BioShop Canada | AGR001 | |
| Soda Lime Galss Beads | Biospec Products | 11079105 | |
| Sodium Chloride | BioShop Canada | SLD002 |
References
- Stagljar, I., Fields, S. Analysis of membrane protein interactions using yeast-based technologies. Trends Biochem Sci. 27 (11), 559-563 (2002).
- Iyer, K. Utilizing the split-ubiquitin membrane yeast two-hybrid system to identify protein-protein interactions of integral membrane proteins. Sci STKE. 275, pl3-pl3 (2005).
- Paumi, C. M. Mapping protein-protein interactions for the yeast ABC transporter Ycf1p by integrated split-ubiquitin membrane yeast two-hybrid analysis. Mol Cell. 26 (1), 15-25 (2007).
- Stagljar, I. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A. 95 (9), 5187-5192 (1998).
- Gisler, S. M. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 7 (7), 1362-1377 (2008).
- Scheper, W. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J Biol Chem. 278 (39), 37998-38003 (2003).
- Thaminy, S. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 13 (7), 1744-1753 (2003).
- Johnsson, N., Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 91 (22), 10340-10344 (1994).
- Kelleher, D. J., Gilmore, R. The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J Biol Chem. 269 (17), 12908-12917 (1994).
- Chevallier, M. R. Cloning and transcriptional control of a eucaryotic permease gene. Mol Cell Biol. 2 (8), 977-984 (1982).
