

Summary
Viene presentata e discussa la formazione di fasci di actomiosina in vitro e la misurazione della generazione della forza dell'insieme di miosina utilizzando pinzette ottiche.
Abstract
Le miosine sono proteine motorie che idrolizzano l'ATP per camminare lungo le tracce del filamento di actina (AF) e sono essenziali nei processi cellulari come la motilità e la contrazione muscolare. Per comprendere i loro meccanismi di generazione della forza, la miosina II è stata studiata sia a livello di singola molecola (SM) che come team di motori in vitro utilizzando metodi biofisici come l'intrappolamento ottico.
Questi studi hanno dimostrato che il comportamento di generazione della forza della miosina può differire notevolmente quando si passa dal livello di singola molecola in una disposizione a tre sfere a gruppi di motori che lavorano insieme su una superficie rigida di tallone o di copertura in una disposizione di scorrimento. Tuttavia, queste costruzioni di saggi non consentono di valutare la dinamica di gruppo della miosina all'interno della gerarchia strutturale viscoelastica come farebbero all'interno di una cellula. Abbiamo sviluppato un metodo che utilizza pinzette ottiche per studiare la meccanica della generazione di forza da parte di insiemi di miosina che interagiscono con più filamenti di actina.
Questi fasci di actomiosina facilitano l'indagine in un ambiente gerarchico e conforme che cattura la comunicazione motoria e l'output della forza d'insieme. La natura personalizzabile del test consente di alterare le condizioni sperimentali per capire come le modifiche all'insieme di miosina, al fascio di filamenti di actina o all'ambiente circostante si traducono in diversi output di forza.
Introduction
Le proteine motorie sono essenziali per la vita, convertendo l'energia chimica in lavoro meccanico 1,2,3. I motori della miosina interagiscono con i filamenti di actina prendendo passi lungo i filamenti simili a una traccia, e la dinamica delle reti actina-miosina svolge la contrazione muscolare, la motilità cellulare, l'anello contrattile durante la citochinesi e il movimento del carico all'interno della cellula, tra gli altri compiti essenziali 3,4,5,6,7,8 . Poiché le miosine hanno così tanti ruoli essenziali, il fallimento nella funzionalità della rete miosina-actina può portare allo sviluppo di malattie, come mutazioni nella catena pesante della miosina che causano ipercontrattilità cardiaca nella cardiomiopatia ipertrofica (HCM)9,10,11,12,13,14 . Nella contrazione muscolare, i singoli motori della miosina cooperano tra loro lavorando come un insieme per fornire l'energia meccanica richiesta che esegue lo scorrimento relativo delle AF 4,15,16,17,18. I motori della miosina formano ponti trasversali tra le AF e utilizzano cambiamenti conformazionali dovuti al suo ciclo meccanochimico per spostarsi collettivamente verso l'estremità spinata dei filamenti allineati 17,18,19,20,21.
Lo sviluppo di saggi quantitativi di motilità in vitro a livello SM utilizzando tecniche come l'intrappolamento ottico ha facilitato la raccolta di dettagli senza precedenti su come funzionano i singoli motori della miosina, compresa la misurazione della generazione della forza SM e delle dimensioni dei passi 22,23,24,25,26,27,28,29,30 . Finer et al. hanno sviluppato il saggio di intrappolamento ottico "a tre perle" o "manubri" per sondare la meccanica di generazione della forza dei motori a singola miosina II23,31. Poiché la miosina muscolare II lavora in team per contrarre la fibrillazione atriale ma non è processiva a livello SM, l'orientamento del saggio di intrappolamento ottico ha dovuto essere riorganizzato dal classico approccio a perline legate al motore32. Per formare il test con manubri, sono state utilizzate due trappole ottiche per tenere un AF su un motore di miosina legato a un tallone attaccato al coprifoglio, e la forza emessa dal singolo motore è stata misurata attraverso i movimenti dell'AF all'interno della trappola23.
Tuttavia, le forze SM e l'utilizzo di un singolo motore / singolo filamento di orientamento del saggio non forniscono un'immagine completa della generazione di forza a livello di sistema poiché molte proteine motorie, inclusa la miosina II, non funzionano isolatamente e spesso non funzionano come somma delle loro parti 15,16,17,32,33,34,35,36 . Strutture più complesse che includono più di un motore che interagisce con più di un filamento sono necessarie per comprendere meglio la sinergia delle reti di filamenti di miosina e actina 15,32. L'orientamento del saggio con manubri è stato sfruttato per studiare la generazione di piccole forze d'insieme avendo miosine multiple attaccate a una perlina o usando un filamento spesso miosina attaccato a una superficie e consentendo ai motori di interagire con l'AF sospeso 4,23,34,37,38,39,40.
Altri piccoli saggi di ensemble includono un saggio di scorrimento a filamento in vitro in cui i motori della miosina sono rivestiti su una superficie di copertura e un tallone legato a un AF viene utilizzato per sondare la forza generata dal team di motori 4,35,36,38,39,40,41,42,43 . In entrambi i casi, le miosine sono legate a una superficie rigida – perlina o coprislip – e utilizzano una fibrillazione atriale. In questi casi, i motori non sono in grado di muoversi liberamente o comunicare tra loro, né avere miosine rigidamente legate riflette l'ambiente gerarchico e conforme in cui i motori lavorerebbero insieme nel sarcomero32. Studi precedenti hanno suggerito che la miosina II può percepire il suo ambiente e adattarsi di conseguenza alle mutevoli condizioni di concentrazione viscoelastica o motoria alterando caratteristiche come la generazione di forza e il rapporto di lavoro41,44,45. Pertanto, è necessario sviluppare un saggio di intrappolamento ottico che promuova e catturi la comunicazione motoria e la conformità del sistema per dipingere un quadro più realistico delle basi meccanicistiche della generazione della forza dell'insieme di miosina II.
Qui, abbiamo sviluppato un metodo per accoppiare la struttura gerarchica in vitro con l'intrappolamento ottico formando fasci di actomiosina o sandwich costituiti da più motori di miosina che interagiscono tra due filamenti di actina. Questa geometria di saggio modulare ha la capacità di sondare direttamente come i fattori molecolari e ambientali influenzano la generazione della forza della miosina dell'insieme. Inoltre, studiare i meccanismi di generazione della forza attraverso questi insiemi di actina-miosina ha il potenziale per aiutare a modellare e comprendere come i compiti cellulari su larga scala, come la contrazione muscolare, si propagano dal livello molecolare 9,10,13.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Copertine per incisione
- Sciogliere 100 g di KOH in 300 ml di etanolo al 100% in un becher da 1.000 ml. Mescolare con una barra di mescolamento fino a quando la maggior parte del KOH si è sciolta.
ATTENZIONE: La soluzione concentrata di KOH può causare ustioni e danni agli indumenti. Indossare guanti, protezioni per gli occhi e un camice da laboratorio. - Posizionare i coprivetrini singolarmente negli scaffali per la pulizia dei coprifogli.
NOTA: I rack sono progettati con fessure che contengono singoli vetrini distanziati per consentire l'incisione e il risciacquo su ciascuna faccia del coprivetrino, i fori di scarico sul fondo e realizzati in materiale in grado di resistere alle difficili condizioni di incisione. Possono essere realizzati su misura o acquistati commercialmente. - Preparare ed etichettare tre becher da 1.000 ml: uno con 300 ml di etanolo e due becher con 300 ml di acqua ad osmosi inversa (RO).
NOTA: Qui, l'acqua RO proviene da un depuratore d'acqua di laboratorio, ma potrebbe anche essere acquistata commercialmente se non è disponibile un depuratore locale. - Posizionare ciascuno dei quattro becher in un sonicatore da bagno per degassare per 5 minuti.
- Immergere un rack di coprivetrini nel becher di KOH ed etanolo e sonicare per 5 minuti.
- Trasferire il rack dei vetrini di copertura dal becher KOH/etanolo al becher di solo etanolo. Immergere il rack su e giù nel becher fino a quando non c'è perline.
NOTA: Fare attenzione a non disturbare i coperchi o a far cadere con forza il rack nel becher. Ciò causerà la fuoriuscita dei coperchi dal rack o causerà schizzi chimici. - Trasferire con cautela il rack di coperchi dal becher di etanolo a un becher d'acqua, immergendo su e giù fino a quando non c'è perline.
- Immergere il rack di coperchi nel becher d'acqua che non è stato ancora utilizzato e sonicare di nuovo per 5 minuti.
- Utilizzare una bottiglia per spruzzare il rack di coprivetrini con acqua fino a quando non scorre senza intoppi dai coprivetrini. Ripetere con l'etanolo.
- Mettere le griglie ad asciugare in forno a 90 °C per 20 minuti. Conservare i rack dei vetrini incisi a temperatura ambiente in contenitori chiusi per evitare contaminazioni prima dell'uso.
2. Polimerizzazione del filamento di actina
- Crea soluzione T
- In un tubo conico da 50 ml, aggiungere 3,94 g di Tris-HCl e 0,147 g di CaCl2. Aggiungere acqua RO per ottenere un volume totale di 50 ml e mescolare bene.
NOTA: Le concentrazioni finali della Soluzione T sono rispettivamente 500 mM Tris-HCl e 20 mM CaCl2. - Etichettare il tubo Soluzione T e conservarlo a 4 °C.
- In un tubo conico da 50 ml, aggiungere 3,94 g di Tris-HCl e 0,147 g di CaCl2. Aggiungere acqua RO per ottenere un volume totale di 50 ml e mescolare bene.
- Crea buffer TC
- Mescolare 40 mL di acqua RO e 1,5 mL di Soluzione T in un tubo conico da 50 mL. Modificare il pH a 8,0 aggiungendo piccole quantità di KOH concentrato. Aggiungere acqua per ottenere 50 ml della soluzione e verificare il pH. Regolare il pH se necessario.
NOTA: Il tampone TC finale contiene 5 mM di Tris-HCl e 0,2 mM di CaCl2 a pH 8. - Etichettare il tubo TC e conservarlo a 4 °C.
- Mescolare 40 mL di acqua RO e 1,5 mL di Soluzione T in un tubo conico da 50 mL. Modificare il pH a 8,0 aggiungendo piccole quantità di KOH concentrato. Aggiungere acqua per ottenere 50 ml della soluzione e verificare il pH. Regolare il pH se necessario.
- Crea buffer FC
- Aggiungere 85 ml di acqua RO, 10 ml di soluzione T, 3,73 g di KCl e 0,041 g di MgCl2 in un flacone tampone da 100 ml. Modificare il pH a 7,5 aggiungendo piccoli volumi di KOH concentrato. Aggiungere acqua per ottenere un volume finale di 100 ml e verificare il pH.
NOTA: il buffer FC finale contiene 500 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2 e 2 mM CaCl2 a pH 7,5. - Etichettare il tubo FC e conservarlo a 4 °C.
- Aggiungere 85 ml di acqua RO, 10 ml di soluzione T, 3,73 g di KCl e 0,041 g di MgCl2 in un flacone tampone da 100 ml. Modificare il pH a 7,5 aggiungendo piccoli volumi di KOH concentrato. Aggiungere acqua per ottenere un volume finale di 100 ml e verificare il pH.
- Preparare il tampone generale di actina (GAB).
- Miscelare 485 μL di tampone TC, 10 μL di 10 mM ATP e 5 μL di 50 mM DTT in una provetta da microcentrifuga.
NOTA: le condizioni finali del buffer sono 5 mM Tris-HCl, 0,2 mM CaCl 2, 0,5 mM DTT e0,2 mM ATP. - Etichettarlo come GAB e conservarlo a 4 °C.
- Miscelare 485 μL di tampone TC, 10 μL di 10 mM ATP e 5 μL di 50 mM DTT in una provetta da microcentrifuga.
- Preparare il tampone di polimerizzazione dell'actina (APB).
- Miscelare 455 μL di tampone FC, 25 μL di 100 mM di ATP e 20 μL di 50 mM DTT in una provetta da microcentrifuga.
NOTA: Le condizioni tampone finali sono 50 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2, 2 mM CaCl2 2 mM DTT e 5 mM ATP. - Etichettare il tubo come APB e conservarlo a 4 °C.
- Miscelare 455 μL di tampone FC, 25 μL di 100 mM di ATP e 20 μL di 50 mM DTT in una provetta da microcentrifuga.
- Ricostituire l'actina
- Ricostituire l'actina del muscolo scheletrico di coniglio aggiungendo 100 μL di acqua deionizzata a un flaconcino da 1 mg di actina liofilizzata. Mescolare bene pipettando delicatamente su e giù. Aliquote in campioni da 5 μL, congelare a scatto e conservare le aliquote di actina da 10 mg/mL a -80 °C.
- Ricostituire l'actina del muscolo scheletrico di coniglio biotinilato aggiungendo 20 μL di acqua RO. Aliquote in campioni da 5 μL, congelare a scatto e conservare le aliquote di actina biotinilata da 1 mg/mL a -80 °C.
- Polimerizzazione dell'actina non marcata con stabilizzazione della rodamina falloidina
- Scongelare un flaconcino da 10 mg/mL di actina e tenerlo in ghiaccio.
- Preparare il tampone GAB fresco, aggiungere 100 μL di GAB all'aliquota di actina e mescolare pipettando delicatamente su e giù. Incubare la soluzione sul ghiaccio per 1 ora.
- Preparare APB fresco durante l'incubazione. Dopo l'incubazione, polimerizzare l'actina in filamenti aggiungendo 11 μL di APB alla soluzione di actina. Mescolare bene pipettando delicatamente su e giù. Mettere in ghiaccio per 20 min.
- Aggiungere 5 μL di falloidina marcata con rodamina alla soluzione di filamento di actina appena polimerizzata. Lasciare sul ghiaccio al buio per 1 ora.
- Conservare il flaconcino di rodamina actina avvolto in un foglio di alluminio al buio a 4 °C.
NOTA: Si consiglia di utilizzare questi filamenti per un periodo massimo di 1 settimana. La qualità della fibrillazione atriale può essere confermata ogni giorno attraverso una rapida immagine di una cella di flusso contenente solo AF e visualizzando filamenti coerenti giorno per giorno.
- Polimerizzazione di actina biotinilata con stabilizzazione della falloidina Alexa Fluor 488
- Scongelare un flaconcino di actina da 10 mg/mL e 1 flaconcino di actina biotinilata da 1 mg/mL e tenerli sotto ghiaccio.
- Crea un nuovo buffer GAB.
- Unire i due flaconcini (punto 2.8.1) in un rapporto actina:actina biotinilata 10:1. Aggiungere 100 μL di GAB alla miscela di actina e mescolare bene pipettando delicatamente su e giù. Incubare su ghiaccio per 1 ora.
- Fai APB fresco durante l'incubazione.
- Dopo la fase di incubazione, polimerizzare l'actina aggiungendo 11 μL di APB alla soluzione di actina. Mescolare bene pipettando su e giù delicatamente. Incubare su ghiaccio per 20 min.
- Aggiungere 5 μL di falloidina marcata con Alexa Fluor 488 e incubare sul ghiaccio al buio per 1 ora.
- Conservare il flaconcino di actina biotinilata avvolto in un foglio di alluminio al buio a 4 °C.
NOTA: Questi filamenti possono essere utilizzati per un periodo massimo di 1 settimana.
3. Preparazione della miosina e delle perline
- Ricostituire la miosina II
- Ruotare brevemente verso il basso (~ 5 s) miosina scheletrica II liofilizzata per raccoglierla sul fondo del tubo usando una minicentrifuga standard.
- Ricostituire la miosina a 10 mg/ml aggiungendo 100 μL di 1 mM DTT preparato in acqua RO.
- Diluire la soluzione madre di miosina 10x aggiungendo 10 μL di 10 mg/mL di miosina a 90 μL di 1 mM DTT in acqua RO. Produrre aliquote di piccolo volume (1-5 μL), congelare a scatto e conservare a -80 °C.
NOTA: L'attività della miosina può essere confermata eseguendo un saggio standard del filamento di scorrimento come pubblicato in precedenza46,47. Vedere la discussione per una breve descrizione.
- Pulizia delle perle rivestite di streptavidina
- Diluire 20 μL di sfere di streptavidina da 1 μm in 80 μL di acqua RO. Lavare quattro volte centrifugando a 9.600 × g e ricostituendo in 100 μL di acqua RO.
- Sonicare per 2 minuti al 40% di ampiezza e conservare le perle lavate su un rotatore a 4 °C.
4. Preparazione della cella di flusso
- Preparare una soluzione di poli-l-lisina (PLL) aggiungendo 30 ml di etanolo al 100% in un tubo da 50 ml e aggiungendo 200 μL di 0,1% p/v di poli-l-lisina in acqua e mescolare bene.
- Aggiungere un coprislip inciso alla soluzione PLL e lasciarlo in ammollo per 15 minuti. Rimuovere il coprislip con una pinzetta, avendo cura di toccare solo il bordo del coprivetrino mentre viene tirato verso l'alto dal tubo (vedi figura 1A-C). Afferrare le copertine per i bordi con una mano guantata.
- Asciugare il coprislip con una linea filtrata fino a quando non rimane etanolo e nessun residuo sul coprifoglio.
- Applicare due pezzi di nastro adesivo biadesivo al centro di un vetrino da microscopio, a 3-4 mm di distanza l'uno dall'altro. Strappare o tagliare il nastro in eccesso che pende dal bordo della diapositiva.
- Aggiungere il coprislip rivestito in PLL sulla parte superiore del nastro perpendicolare all'asse lungo del vetrino del microscopio (formando una T) per formare un canale.
- Utilizzare un tubicino per comprimere il vetrino sul nastro e il vetrino del microscopio accuratamente fino a quando il nastro non è trasparente (Figura 1A). Assicurarsi che non vi siano bolle nel nastro in quanto ciò può causare perdite dal canale di flusso.
NOTA: La cella di flusso può contenere un volume di 10-15 μL.
5. Preparazione del fascio di actiomiosina
- In provette separate, diluire ogni tipo di filamento di actina (marcato con rodamina e biotinilato 488) 600x mescolando 0,5 μL della relativa actina marcata con 300 μL di APB. Aggiungere altri 5 μL della falloidina marcata in modo corrispondente a ciascuna provetta e incubare sul ghiaccio al buio per 15 minuti.
- Alla soluzione di actina biotinilata, aggiungere un sistema di evacuazione dell'ossigeno di 1 μL di beta-D-glucosio a 500 mg / ml, 1 μL di glucosio ossidasi a 25 mg / ml e 1 μL di catalasi a 500 unità / ml. Aggiungere 1 μL di 100 mM di ATP e 1 μL di perle di streptavidina diluite e pulite 100x. Mescolare delicatamente con una punta di pipetta. Mettere la sospensione su un rotatore a 4 °C mentre il resto del fascio di actomiosina viene assemblato.
- Aggiungere 15 μL di rodamina actina diluita alla cella di flusso PLL (Figura 1D). Stoppare la soluzione in eccesso attraverso la cella di flusso, ma non lasciare che il canale di flusso si asciughi. Incubare per 10 minuti in una camera di umidità.
NOTA: Le camere di umidità possono essere realizzate con scatole di punta per pipette vuote con acqua aggiunta sul fondo e coperchio coperto in foglio di alluminio per bloccare la luce. - Preparare una soluzione di caseina da 1 mg/mL in APB.
- Aggiungere 15 μL di caseina da 1 mg/mL per evitare il legame non specifico dei componenti successivi (Figura 1E). Incubare per 5 minuti in una camera di umidità.
- Aggiungere la concentrazione desiderata di miosina all'actina biotinilata e alla sospensione di perline dal punto 5.2. Mescolare delicatamente con la punta della pipetta, quindi aggiungere immediatamente 15 μL della sospensione del passo 5.2 + la concentrazione di miosina desiderata alla cella di flusso (Figura 1F,G). Incubare per 20 min. Sigillare le estremità aperte della cella di flusso con smalto per unghie per evitare l'evaporazione durante gli esperimenti di imaging e di intrappolamento ottico.
NOTA: Una concentrazione di soluzione di miosina di 1 μM produce un raggruppamento robusto e può essere utilizzata come punto di partenza per la personalizzazione desiderata del saggio (vedere Figura 2).
6. Misurazioni della forza utilizzando trappola ottica (NT2 Nanotracker2)
NOTA: mentre il protocollo riportato di seguito è specifico per il sistema NT2, questo test può essere utilizzato con altri sistemi di intrappolamento ottico, compresi quelli costruiti su misura, che hanno anche capacità di fluorescenza. Il flusso di lavoro generale rimane lo stesso di mettere a fuoco la superficie del vetrino, eseguire calibrazioni di perline e acquisire dati trovando fasci di actina fluorescenti. Per il sistema NT2, la figura supplementare S1, la figura supplementare S2, la figura supplementare S3, la figura supplementare S4, la figura supplementare S5, la figura supplementare S6 e la figura supplementare S7 forniscono dettagli sul sistema di intrappolamento ottico e sull'interfaccia software.
- Accendere la scatola di controllo e il laser (Figura supplementare S1).
- Avviare il software del computer trappola ottica facendo clic sull'icona JPK Nanotracker sul desktop.
- Riattivare il telecomando facendo clic sul pulsante Logitech al centro (figura supplementare S2).
- Accendere il modulo di fluorescenza attivando o disattivando l'interruttore (Figura supplementare S3).
- Ruotare la torretta del cubo del filtro per l'imaging in campo chiaro (Figura supplementare S4).
- Una volta che il sistema è pronto, accendere il laser utilizzando il pulsante di accensione laser nell'angolo in basso a sinistra dello schermo a 50 mW e lasciarlo stabilizzare per 30 minuti (Figura supplementare S5).
- Fare clic in sequenza sui pulsanti Illuminazione, Fotocamera, Obiettivo e Movimento palcoscenico all'interno del software per visualizzare e modificare tali finestre durante l'esperimento. Accendere l'illuminazione del microscopio facendo clic sul pulsante On/Off e impostandolo sulla massima potenza facendo clic e trascinando la barra completamente verso destra (Figura supplementare S5).
- Aprire l'area del campione e rimuovere il supporto del campione dallo stadio del microscopio. Aggiungere la cella di flusso, fissarla con i portacampioni metallici e assicurarsi che il vetrino con il coprislip sia sul fondo.
- Aggiungere 30 μL di acqua RO al centro dell'obiettivo inferiore. Non lasciare che la punta della pipetta tocchi l'obiettivo. Reinserire la fase di esempio.
NOTA: poiché il sistema NT2 utilizza un obiettivo di immersione in acqua come obiettivo di intrappolamento, il supporto di immersione può essere diverso a seconda dell'obiettivo di abbondanza nella configurazione dell'utente. - Sollevare l'obiettivo inferiore utilizzando le frecce di controllo sullo schermo o L2 sul telecomando fino a quando il tallone d'acqua tocca il coprislip (Figura supplementare S5).
- Abbassare l'obiettivo superiore fino a raggiungere circa la metà della distanza dalla cella di flusso utilizzando le frecce sullo schermo o R2 sul telecomando. Aggiungere 170 μL di acqua RO alla parte superiore della cella di flusso direttamente sotto l'obiettivo superiore. Abbassare l'obiettivo superiore fino a quando non rompe la tensione superficiale dell'acqua e forma un menisco.
- Spostare lo stadio del microscopio utilizzando il pad a freccia sul telecomando fino a raggiungere il bordo del nastro adiacente al canale di flusso. Chiudere lo sportello del campione.
NOTA: un "clic" alla chiusura dello sportello del campione indica che l'otturatore laser è ora aperto. Questa è una funzione di sicurezza che consente l'apertura della tapparella solo se la porta è chiusa. - Utilizzando la finestra Obiettivo sullo schermo, mettere a fuoco il bordo del nastro spostando verso l'alto l'obiettivo inferiore denominato Obiettivo laser facendo clic sulla freccia superiore utilizzando i controlli sullo schermo. Fate lo stesso per l'obiettivo superiore facendo clic sulla freccia inferiore (Figura supplementare S5).
NOTA: le doppie frecce spostano l'obiettivo o lo stage più velocemente. Il bordo del nastro viene utilizzato per la messa a fuoco perché è un oggetto grande e facile da trovare che si trova vicino alla superficie del coprifoglio. Le bolle d'aria all'interno del nastro sono un'altra opzione. Tuttavia, questa operazione non è necessaria se l'utente dispone di una routine automatizzata per trovare lo stato attivo della superficie o di un metodo interno preferito. - Una volta che il nastro è a fuoco, chiudere parzialmente il diaframma nella parte superiore della trappola ottica. Abbassa l'obiettivo superiore fino a quando non è visibile la forma poligonale dell'iride. Mettere a fuoco questi bordi, riaprire il diaframma, quindi accoppiare gli obiettivi facendo clic sull'icona del lucchetto (Figura supplementare S5).
- Trova una perlina galleggiante e intrappolala facendo clic sul pulsante Trap Shutter , che aprirà l'otturatore e consentirà al laser di intrappolare il campione. Fare clic sul cursore Abbondanza sullo schermo e trascinarlo per spostare la posizione del laser di abbondanza . Una volta intrappolato, calibrare il tallone per correlare le misurazioni della tensione alla forza e allo spostamento.
- Fare clic sul pulsante Calibrazione . Regolare la routine di calibrazione in base all'analisi degli spettri di potenza e adattare la frequenza d'angolo all'interno del software per le direzioni X, Y e Z (Figura supplementare S6).
- Fare clic su Impostazioni. Digitare il diametro del tallone (1.000 nm) e digitare la temperatura dello stadio che si trova in basso a sinistra della finestra del software. (cfr . figura supplementare S6).
- Fare clic su Trap 1. Fare clic su Segnale X. Fare clic su Esegui per eseguire l'adattamento della frequenza d'angolo. Fare clic e trascinare all'interno della finestra per ottimizzare la funzione di adattamento. Fare clic su Usalo per i valori di sensibilità e rigidità. Fare clic su Accetta valori. Ripetere l'operazione per i segnali Y e Z. Chiudere la finestra. (cfr . figura supplementare S6).
NOTA: Le routine di calibrazione del tallone su altri sistemi di intrappolamento ottico o sistemi personalizzati che sono stati testati in modo robusto dall'utente, come il metodo di equipartizione o il metodo della forza di trascinamento, sono accettabili anche57,58. - Trova un fascio di actomiosina cercando perline legate alle AF sulla superficie del vetrino.
- Quando viene rilevata una perla non affollata da altre perle galleggianti, osservare gli AF intorno ad essa mediante imaging a fluorescenza per verificare la presenza di un fascio.
- Verificare che sia presente un fascio cercando entrambi gli AF fluorescenti colocalizzati. Accendere la sorgente di luce bianca e utilizzare il cubo filtrante appropriato per visualizzare ciascun filamento di actina ruotando la torretta (cubi del filtro di eccitazione 488 nm e 532 nm per l'eccitazione Alexa Fluor 488 e rodamina, rispettivamente). Vedere la figura supplementare S4.
NOTA: Un esperimento di controllo per verificare l'intensità di fluorescenza di singoli AF può essere utile per identificare fasci composti da un singolo filamento marcato con rodamina 488 e singolo, o applicabile a qualsiasi set di fluorofori che l'utente sceglie di utilizzare. - Una volta verificato, intrappolare il tallone attaccato al filamento superiore del fascio facendo clic sul pulsante Trap Shutter .
- Utilizzare i controlli sullo schermo per registrare i dati facendo clic sul pulsante Oscilloscopio (Figura supplementare S7). Per visualizzare le misurazioni senza registrare i dati, fare clic su Start. Per salvare tutti i dati, fare clic su Salvataggio automatico. Per registrare le misurazioni, fare clic su Avvia registrazione. Scegli quali dati devono essere visualizzati in tempo reale (posizione, forza, direzione x, direzione y) scegliendo dal menu a discesa Segnale X o segnale Y. Ricorda che xdirection è da sinistra a destra e la direzione y è su e giù sullo schermo. Vedere la figura supplementare S7.
NOTA: i dati verranno salvati come file .out e includono tempo, tensione, spostamento e forza per ogni direzione. Questi file possono essere esportati in altri software per la visualizzazione e l'analisi.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Le celle a flusso contenenti i sistemi di fasci di actomiosina hanno un design standard, costituito da un vetrino da microscopio e da un coprivetrino inciso separati da un canale costituito da nastro adesivo biadesivo (Figura 1). Il test viene quindi costruito dal coverslip up utilizzando introduzioni graduali come descritto nel protocollo. Il test finale consiste in filamenti di actina marcati con rodamina; la concentrazione di miosina desiderata (1 μM è stato utilizzato per i risultati rappresentativi nelle figure 2 e 3); filamenti di actina biotinilati, marcati con Alexa Fluor 488; 1 μm di perle di streptavidina; il sistema di evacuazione dell'ossigeno; ATP; e buffer APB. Si formeranno più fasci per cella di flusso e le concentrazioni di actina sopra descritte forniscono un'adeguata spaziatura tra i fasci per garantire che non ci siano interazioni indesiderate. Ciò facilita anche l'ottenimento di misurazioni multiple della forza per cella di flusso per aumentare l'efficienza di acquisizione dei dati. I profili di forza dovrebbero essere riproducibili all'interno di una cella di flusso e da cella di flusso a cella di flusso.
Mentre il protocollo di cui sopra è orientato verso l'uso di una configurazione di trappola ottica commerciale, la cella di flusso e il saggio presentati qui potrebbero essere facilmente utilizzati per un diverso strumento commerciale o una configurazione di trappola ottica personalizzata accoppiata con un microscopio o uno stadio di microscopio e in possesso di capacità di imaging a fluorescenza. Una volta completate tutte le aggiunte di celle di flusso secondo il protocollo di cui sopra, i fasci di actomiosina sul vetrino (Figura 1) sono pronti per la misurazione immediata. La cella di flusso viene aggiunta allo stadio del microscopio ottico trappola, vengono acquisite più misure di calibrazione delle perle e i fasci vengono identificati attraverso la colocalizzazione a fluorescenza dei filamenti del fascio. Una perlina legata a un fascio viene intrappolata e inizia lo spostamento e la misurazione della forza corrispondente. L'utente può osservare l'acquisizione dei dati in tempo reale sul monitor del computer. A seconda della concentrazione di miosina utilizzata nella cellula a flusso, il fascio potrebbe iniziare a mostrare immediatamente un movimento sostanziale, oppure potrebbero essere necessari 30 s-1 min per vedere efficacemente un aumento di spostamento / forza.
Una traccia di forza rappresentativa è mostrata nella Figura 3A dove i motori della miosina mostrano una rampa costante di forza seguita da un plateau. È tipico vedere questi tipi di tracce svilupparsi nell'arco di 2-5 minuti. Tuttavia, è anche possibile misurare i fasci di attomiosina che non generano alcuna forza netta (Figura 3B). Queste tracce appaiono come rumore di base o non presentano un aumento netto sostanziale della forza oltre i 90 s. Ciò è probabilmente dovuto a una bassa concentrazione locale del motore che non consente uno scorrimento produttivo, o il fascio è in un orientamento parallelo sfavorevole in cui le estremità più e meno dei filamenti sono allineate.
Poiché il contenuto della cella di flusso può essere suscettibile alla degradazione dall'illuminazione incidente e dal laser di intrappolamento, al riscaldamento locale sul vetrino nel tempo e alla generazione di specie di ossigeno radicale, si consiglia vivamente di non utilizzare la stessa cella di flusso per più di 1 ora. Per la massima efficienza, si suggerisce di avere un altro test in incubazione durante l'acquisizione dei dati. La traccia di spostamento/forza può essere esportata dal software di trappola ottica in Excel, Matlab, Igor o altri programmi di gestione dei dati per ulteriori filtri e analisi. I dati che possono essere estratti da tali esperimenti di intrappolamento ottico di ensemble/bundle includono diversi tipi di profili di generazione della forza (linea di base, rampa/plateau) in diverse condizioni di analisi, velocità di generazione della forza, generazione di forza massima, comportamento cinetico dell'insieme e del passo attraverso le dimensioni dei passi e i tempi di permanenza tra i passi o le squadre di passi, nonché il rapporto di lavoro. L'utente può anche modificare le condizioni del test per confrontare il modo in cui l'aggiunta di diversi tipi di motori della miosina, l'aggiunta di proteine leganti l'actina o la modifica delle condizioni tampone influenzano queste caratteristiche di generazione della forza dell'insieme.
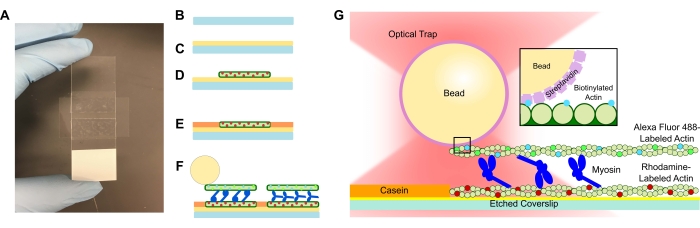
Figura 1: Schema del saggio. (A-C) I vetrini incisi sono rivestiti in poli-L-lisina e utilizzati per formare la cella di flusso utilizzando nastro biadesivo e un vetrino da microscopio. Le introduzioni temporizzate, le fasi di incubazione descritte nel protocollo si traducono in actina stabilizzata con falloidina marcata con rodamina come modello o filamento inferiore (D), seguita dal blocco della caseina per prevenire il legame non specifico (E) e (F) Alexa Fluor 488 actina biotinilata stabilizzata con falloidina come carico o filamento superiore e squadre di miosina II che fanno scorrere i filamenti e generano forza quando viene introdotto ATP. La geometria dei motori e la natura della reticolazione all'interno del fascio potrebbero variare in condizioni diverse, come la concentrazione salina59. Studi precedenti hanno dimostrato che il dominio della coda della miosina ha la capacità di interagire con i filamenti di actina e la lenta motilità dell'ensemble46. Tuttavia, le teste di miosina in esperimenti pesanti di meromiosina dimostrano il legame di ciascuna testa ai filamenti di actina adiacenti60. (G) Le sfere di streptavidina sono utilizzate come impugnatura ottica per la trappola e si legano esclusivamente al filamento di actina biotinilata del carico, il che aiuta a convalidare la formazione di fasci appropriati sul vetrino. Fare clic qui per visualizzare una versione ingrandita di questa figura.

Figura 2: Fasci fluorescenti di actomiosina. Quattro diversi incontri di filamenti di actina e fasci all'interno del saggio del fascio presentato in Figura 1. Il filamento di actina biotinilata cargo superiore con il canale della falloidina Alexa Fluor 488 è mostrato a sinistra e il filamento di actina del modello inferiore con il canale della falloidina della rodamina è sulla destra. In basso, la stessa figura è mostrata con linee colorate sovrapposte per aiutare a guidare l'occhio. (A) Un filamento di actina superiore si trova vicino a un filamento di actina inferiore ma ha una sovrapposizione incompleta. Questo non verrebbe utilizzato per gli esperimenti bundle. (B) I filamenti di actina superiore e inferiore sono colocalizzati e l'intensità di ciascun filamento conferma che si tratta di ogni singolo filamento all'interno del fascio. Questo sarebbe un buon candidato per gli esperimenti di bundle. (C) Un grande fascio di filamenti di rodamina autoassemblati si trova sul fondo. Mentre c'è un corrispondente filamento di actina superiore che è colocalizzato, ci sono troppi filamenti inferiori presenti; Pertanto, non verrebbe utilizzato per esperimenti di bundle. Questo è anche un esempio di come quando più filamenti di actina dello stesso tipo sono raggruppati, l'intensità della fluorescenza aumenta. L'utente può utilizzarlo come misuratore per giudicare singoli filamenti rispetto a fasci dello stesso tipo di filamento. (D) È presente un filamento inferiore senza filamento superiore corrispondente, confermando anche l'assenza di sanguinamento. Questo non verrebbe utilizzato per gli esperimenti bundle. Notiamo che l'intensità dei filamenti nel canale Alexa Fluor 488 è bassa e crediamo che sia dovuta al set di filtri che viene utilizzato (Set di filtri 09 di Zeiss). Il set di filtri utilizzato per il canale della rodamina è il set di filtri 43 di Zeiss. Fare clic qui per visualizzare una versione ingrandita di questa figura.

Figura 3: Generazione della forza d'insieme di Myosin II. Tracce rappresentative di motori scheletrici della miosina II che generano forza all'interno della gerarchia strutturale dell'actina costruita in vitro . I motori della miosina lavorano insieme per generare collettivamente e produttivamente forza fino a raggiungere un plateau e la forza è sostenuta (A) o sperimentare l'antagonizzazione vicino alla linea di base (B). Fare clic qui per visualizzare una versione ingrandita di questa figura.
Figura supplementare S1: trappola ottica Bruker/JPK Nanotracker2. (A) Monitor del computer. (B) Tastiera del computer. (C) Torre di computer. (D) Scatola del controller. (E) Alimentazione laser. (F) Scatola ottica trappola ottica. (G) Microscopio invertito. (H) Porta allo stadio del microscopio. (I) Cursore polarizzatore per passare dall'imaging a contrasto a campo chiaro e interferenza differenziale. Clicca qui per scaricare questo file.
Figura supplementare S2: Telecomando per trappola ottica. (A) Tastiera per posizionare il palco motorizzato. (B-C) Regolare la posizione dell'abbondanza. (D) A, X e B accendono e disattivano rispettivamente l'otturatore principale, l'otturatore dell'otturatore dell'abbondanza 1 e l'otturatore dell'otturatore dell'abbondanza 2. (E) Il pulsante Logitech viene utilizzato per riattivare il controller. (F) I pulsanti su e giù utilizzati per posizionare l'obiettivo di abbondanza. (G) I pulsanti su e giù utilizzati per posizionare l'obiettivo di rilevamento. Si noti che il telecomando non è richiesto e tutte queste manipolazioni possono essere eseguite nel software. Tuttavia, è conveniente essere in grado di controllare gli obiettivi e la posizione del palcoscenico mentre si guarda nell'ambiente del palcoscenico del microscopio. Clicca qui per scaricare questo file.
Figura supplementare S3: Modulo a fluorescenza per trappola ottica. La sorgente di luce bianca a fluorescenza 89North PhotoFluor è accoppiata al retro del microscopio invertito. Si accende e si spegne con un interruttore a levetta (freccia). Clicca qui per scaricare questo file.
Figura supplementare S4: Torretta cubica con filtro a fluorescenza. La torretta (freccia) può essere ruotata per utilizzare il cubo del filtro necessario per l'imaging nei coloranti DIC, rhodamine o Alexa Fluor 488. Si noti che i cubi del filtro possono essere disattivati per personalizzare la configurazione per l'utilizzo di diversi fluorofori. Clicca qui per scaricare questo file.
Figura supplementare S5: software Nanotracker2. (A) Pulsante di accensione e controllo laser. (B) Finestra di posizionamento dell'obiettivo. Le frecce direzionali vengono utilizzate per spostare gli obiettivi di rilevamento (in alto) e di abbondanza (in basso). Le doppie frecce spostano gli obiettivi a una velocità maggiore. Il pulsante blu e rosso in basso a sinistra disaccoppia gli obiettivi e li riporta alla loro posizione originale. Questo è necessario per quando si prelevano campioni dentro e fuori dalla fase del microscopio. Il terzo pulsante da sinistra con gli obiettivi e l'icona del lucchetto "accoppia" gli obiettivi in modo che quando sono entrambi a fuoco e raggiungono l'illuminazione Kohler, l'utente può spostare sia gli obiettivi di abbondanza che di rilevamento su e giù nell'asse z. (C) Finestra di posizionamento del campione utilizzata per spostare lo stadio del microscopio sugli assi x e y. Le doppie frecce spostano il palcoscenico a una velocità maggiore. Questa finestra viene attivata facendo clic sull'icona della freccia su/giù e sinistra/destra nel menu in alto. (D) Finestra di visualizzazione della fotocamera. L'icona della chiave inglese può essere utilizzata per impostare condizioni di imaging personalizzate. Questa finestra si attiva facendo clic sull'icona Fotocamera nel menu in alto. (E) Finestra di illuminazione del microscopio. Questa finestra si attiva cliccando sull'icona Lampadina nel menu in alto. Clicca qui per scaricare questo file.
Figura supplementare S6: Finestra di calibrazione. (A) Questa finestra viene utilizzata per la calibrazione delle perline e si attiva facendo clic sull'icona Cal nel menu in alto. Per calibrare un tallone, si ottiene un adattamento ottimale della frequenza d'angolo nei segnali x, y e z. (B) Per ogni segnale, scegliere il pulsante di segnale appropriato in alto a sinistra. (C) Fare clic su Esegui e ottimizzare l'adattamento facendo clic e trascinando all'interno della finestra verde (D). (E) Una volta soddisfatto dell'adattamento, fare clic su Usalo per sensibilità e rigidità. Ciò consentirà di registrare lo spostamento in nanometri e la forza nei piconewton. (F) Quindi, fai clic su Accetta valori in basso a sinistra. Ripetere l'operazione per le direzioni y e z. Clicca qui per scaricare questo file.
Figura supplementare S7: finestra di acquisizione dati. Questa finestra viene utilizzata per acquisire dati di posizione e forza e consente all'utente di vedere le misurazioni in tempo reale. (A) Questa finestra si attiva facendo clic sull'icona x,t nel menu in alto. (B) L'utente può passare dalla visualizzazione dei segnali x e y. (C) Fare clic su Start per iniziare a visualizzare i dati. Fare clic su Autosave per salvare i dati. Fare clic su Avvia registrazione per iniziare a registrare e salvare i dati. Clicca qui per scaricare questo file.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Uno studio in vitro utilizzando pinzette ottiche combinate con imaging a fluorescenza è stato eseguito per studiare la dinamica degli insiemi di miosina che interagiscono con i filamenti di actina. I fasci di actina-miosina-actina sono stati assemblati utilizzando miosina muscolare II, actina rodamina nella parte inferiore del fascio e sulla superficie del coprislip e filamenti di actina biotinilati marcati con 488 sulla parte superiore del fascio. La proteina actina del muscolo di coniglio è stata polimerizzata e stabilizzata utilizzando tamponi generali di actina (GAB) e tamponi polimerizzanti di actina (APB). GAB e APB devono essere preparati ogni giorno in laboratorio utilizzando ATP, tampone FC e tampone TC. La miosina muscolare II è stata utilizzata per formare i sandwich actina-miosina-actina. La falloidina è stata utilizzata per la colorazione fluorescente dei filamenti di actina e la stabilizzazione in vitro.
L'attività della miosina può essere confermata eseguendo un saggio standard del filamento di scorrimento come pubblicato in precedenza46,47. La miosina II e i suoi sottoframmenti possono legarsi alla superficie del coverslip in una varietà di orientamenti e la presenza del dominio di coda può rallentare lo scorrimento del filamento rispetto ai saggi che utilizzano la meromiosina pesante46,48,49. Tuttavia, il volo a vela e il movimento della superficie possono ancora essere osservati. Una dimostrazione più evidente dell'attività della miosina è la rottura attiva del filamento di actina che può essere osservata dove i filamenti di actina più lunghi vengono rotti in frammenti più piccoli che poi scivolano via in più direzioni. Ciò si verifica a causa dell'alta concentrazione di motori attivi sulla superficie, è stato osservato da più laboratori e non si verifica senza motori miosinci attivi presenti 42,50,51,52,53,54. Inoltre, il test del fascio qui presentato aiuta ad alleviare i problemi di motilità che sono stati principalmente associati al saggio del filamento scorrevole, come la varietà di orientamenti di legame del motore su un vetrino di copertura, perché il test del fascio comporta il blocco della caseina della superficie del vetro in modo che i motori si leghino all'interno del fascio 47,55,56.
Il primo passo è aggiungere filamenti di rodamina actina come filamento inferiore o modello a un coprislip rivestito di poli-L-lisina in una cella di flusso. La poli-L-lisina è usata per promuovere il legame con l'actina poiché la poli-lisina è caricata positivamente mentre l'actina ha cariche negative ed è stata utilizzata in precedenti preparazioni di test citoscheletrici in vitro 61,62,63. Prima della formazione del fascio, diverse diluizioni di actina sono state aggiunte a una cella a flusso per ottimizzare la concentrazione di actina. In questo caso, 600x dal magazzino era la diluizione ottimale che ha prodotto un numero sufficiente di filamenti modello per la formazione del fascio ma con una spaziatura adeguata in modo che i fasci fossero individualizzati. La diluizione è stata effettuata utilizzando il tampone APB. L'aggiunta di rodamina actina è stata seguita da uno strato di caseina per bloccare la superficie ed evitare legami non specifici. La cella di flusso è stata incubata per 30 minuti e lavata dopo l'incubazione con tampone per lavare via eventuali filamenti di actina non legati. Infine, una combinazione di miosina, actina 488/biotina e perle rivestite di streptavidina sono state aggiunte alla cella di flusso per facilitare la formazione del fascio actina-miosina. La concentrazione del tallone dovrebbe essere tale che ce ne siano abbastanza per legare i fasci legati alla superficie e abbastanza in sospensione per facilitare la calibrazione. Tuttavia, una concentrazione troppo elevata di perline può causare difficoltà durante gli esperimenti di intrappolamento a causa delle perle vicine che cadono nella trappola laser e interrompono la misurazione. I motori di miosina vengono aggiunti alla combinazione subito prima di iniettarla nel vetrino in modo che i motori di miosina non si aggreghino preventivamente con il carico o il filamento di actina biotinilata superiore e quindi leghino la rodamina inferiore per fasciare filamenti di actina biotinilata.
Il sistema di trappola ottica NT2 è una trappola ottica commerciale con modalità combinate di campo chiaro, contrasto di interferenza differenziale (DIC) ed epifluorescenza. È accoppiato con un microscopio invertito Zeiss AxioObserver 3 con obiettivi di intrappolamento e rilevamento in acqua 100x/NA 1.46 e 63x/NA 1.0. Il sistema è dotato della capacità di cattura di clic e trascinamento di una trappola laser e può essere utilizzato durante l'imaging in una qualsiasi delle modalità elencate in precedenza. I fasci formati vengono rilevati e confermati utilizzando l'imaging a fluorescenza. Avere una sorgente di luce bianca con cubi filtranti appropriati (GFP / FITC e TRITC / CY3) consente una rapida commutazione tra le immagini del filamento. Gli AF colocalizzati sono stati verificati visualizzando gli AF alle diverse lunghezze d'onda di eccitazione prima di effettuare ogni misurazione della forza utilizzando una pinzetta ottica. Poiché i filamenti possono fotobleggiare rapidamente anche con un reagente di evacuazione dell'ossigeno, si suggerisce ai ricercatori di ottimizzare i parametri di visualizzazione come l'intensità e il tempo di esposizione prima di eseguire gli esperimenti del fascio.
L'intrappolamento ottico è stato impiegato per effettuare le misurazioni della forza, utilizzando le sfere di streptavidina in presenza di ATP per legare il filamento di actina del carico biotinilato e attivare la generazione della forza della miosina come trasduttore di forza. I dati di spostamento e forza rispetto al tempo ottenuti dall'intrappolamento ottico sono stati estratti dal software di intrappolamento per l'analisi. Tuttavia, il software di trappola commerciale fornisce anche routine di analisi che possono essere utilizzate, oppure algoritmi personalizzati in altri programmi possono essere programmati dall'utente per visualizzare e analizzare i dati di trappola. Sui sistemi di trappola ottica personalizzati, l'utente può avere laser di eccitazione invece di una fonte di luce bianca con filtri, che sono anche accettabili da usare. Inoltre, i coloranti fluorescenti possono essere modificati per adattarsi alle apparecchiature esistenti che un utente può avere se gli spettri di emissione non si sovrappongono e causano lo spurgo.
Notiamo che il test presentato è un test di base che può essere ulteriormente personalizzato dall'utente a seconda della sua domanda di ricerca nell'ambito della meccanica dell'ensemble di actomiosina. Il flusso di lavoro generale può anche essere applicato ad altri sistemi di ensemble citoscheletrico in vitro che possono essere di interesse, come i saggi del fascio di microtubuli che formano modelli minimi di fuso mitotico 32,61,63,64,65,66. Le modifiche potrebbero includere, ma non sono limitate alla modifica delle etichette dei fluorofori che sono adatte alla configurazione esistente dell'utente; alterare la concentrazione, il costrutto o l'isotipo della miosina; e la titolazione delle condizioni tampone, tra gli altri aspetti.
Potenziali sfide sono possibili quando si esegue questo test. Quando si formano i fasci actina-miosina, la concentrazione di miosina all'interno dei fasci di actina potrebbe non essere omogenea attraverso il vetrino. Per far fronte a ciò, verranno misurati più fasci sull'intera diapositiva per garantire che i profili di distribuzione del motore e di generazione della forza siano campionati correttamente. È anche difficile conoscere l'orientamento del fascio se questo è necessario per l'interpretazione dei dati di forza. Pertanto, dovrebbero essere prese più prove per ogni pacchetto. Si potrebbe anche incorporare l'etichettatura finale del filamento di actina attraverso gelsolin fluorescente o perline rivestite di gelsolina di dimensioni inferiori rispetto alla maniglia di intrappolamento ottico. L'imaging a fluorescenza può anche essere usato per esaminare le forze delle componenti x e y per dedurre l'orientamento del fascio. Inoltre, poiché lo stato di aggregazione della miosina è fortemente influenzato dalla forza ionica del tampone con formazione di filamenti spessi che si verificano dopo una rapida diluizione di KCl, la concentrazione di sale tampone deve essere monitorata in modo appropriato67,68.
Studi precedenti che utilizzavano altri metodi in vitro come i saggi di scorrimento sono stati utili per identificare il ruolo dei domini della miosina e studiare la configurazione e le interazioni tra la miosina e altre proteine leganti l'actina. Tuttavia, questi metodi hanno uno svantaggio in quanto legare la miosina su una superficie rigida limiterà il potenziale di coordinamento tra i motori della miosina e quindi il feedback meccanosensoriale che si verifica per determinare se l'insieme del motore è in una modalità di rapporto di servizio alto o basso 33,35,41,69. Inoltre, l'intrappolamento ottico con reti motorie a miosina singola non fornisce una chiara comprensione di come i motori della miosina interagiscono tra loro e con i filamenti di actina. Il protocollo sviluppato qui consente lo studio della dinamica dell'insieme motorio della miosina all'interno di una rete di actina conforme e gerarchica. È anche personalizzabile in termini di caratteristiche dell'insieme motore-filamento come concentrazione, isoforma e ambiente tampone, tra gli altri aspetti, per consentire un'indagine sistematica. Il protocollo presentato è una piattaforma per studi futuri di reti di actomiosina più complesse e mantiene la precisione delle misurazioni di spostamento e generazione di forza facilitata dall'intrappolamento ottico che è stato tradizionalmente utilizzato per studi su singole molecole.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno conflitti di interesse da dichiarare.
Acknowledgments
Questo lavoro è supportato in parte dalla University of Mississippi Graduate Student Council Research Fellowship (OA), dall'Università del Mississippi Sally McDonnell-Barksdale Honors College (JCW, JER), dal Mississippi Space Grant Consortium con il numero di sovvenzione NNX15AH78H (JCW, DNR) e dall'American Heart Association con il numero di sovvenzione 848586 (DNR).
Materials
| Name | Company | Catalog Number | Comments |
| Actin protein (biotin): skeletal muscle | Cytoskeleton | AB07-A | Biotinylated actin protein |
| Actin protein, rabbit skeletal muscle | Cytoskeleton | AKL99-A | Actin protein |
| Alexa Fluor 488 Phalloidin | Invitrogen | A12379 | Actin stabilizer and Alexa Fluor 488 stain |
| ATP | Fisher scientific | BP413-25 | Required for actin assembly and myosin motility |
| Beta-D-glucose | Fisher scientific | MP218069110 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Blotting Grade Blocker (casein) | Biorad | 1706404 | Used to block surface from non-specific binding |
| CaCl2 | Fisher scientific | C79500 | Calcium chloride, provides the necessary control over the dynamics of actin myosin network |
| Catalase | Fisher scientific | ICN10040280 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Coverslips | Fisher scientific | 12544C | Used to make flow cells |
| DTT | Fisher scientific | AC327190010 | Used for buffer preparation |
| Ethanol | Fisher scientific | A4094 | Regent used for cleaning coverslips |
| Glucose oxidase | Fisher scientific | 34-538-610KU | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| KCl | Fisher scientific | P217-500 | Used for buffer preparation |
| KOH | Fisher scientific | P250-1 | Used to etch coverslips and adjust buffer pH |
| MgCl2 | Fisher scientific | M33-500 | Used for buffer preparation |
| Microscope slides | Fisher scientific | 12-544-2 | Used to make flow cells |
| Myosin II protein: rabbit skeletal muscle | Cytoskeleton | MY02 | Full length myosin motor protein isolated from rabbit skeletal muscle |
| Nanotracker2 | Bruker/JPK | NT2 | Optical trapping instrument |
| Poly-l-lysine | Sigma-Aldrich | P8920 | Facilities adhesion of actin filaments onto glass surface of the coverslip |
| Rhodamine Phalloidin | Cytoskeleton | PHDR1 | Actin stabilizer and rhodamine fluorescent stain |
| Streptavidin beads, 1 μm | Spherotech | SVP-10-5 | Optical trapping handle |
| Tris-HCl | Fisher scientific | PR H5121 | Used for buffer preparation |
References
- Goldstein, L. S. Kinesin molecular motors: transport pathways, receptors, and human disease. Proceedings of the National Academy of Sciences of the United States of America. 98 (13), 6999-7003 (2001).
- Lee Sweeney, H., Holzbaur, E. L. F.
- O'Connell, C. B., Tyska, M. J., Mooseker, M. S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochimica et Biophysica Acta - Molecular Cell Research. 1773 (5), 615-630 (2007).
- Kaya, M., Tani, Y., Washio, T., Hisada, T., Higuchi, H. Coordinated force generation of skeletal myosins in myofilaments through motor coupling. Nature Communications. 8, 1-13 (2017).
- Akhshi, T. K., Wernike, D., Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 71 (1), 1-23 (2014).
- Brawley, C. M., Rock, R. S. Unconventional myosin traffic in cells reveals a selective actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 106 (24), 9685-9690 (2009).
- Hartman, M. A., Spudich, J. A. The myosin superfamily at a glance. Journal of Cell Science. 125 (7), 1627-1632 (2012).
- Spudich, J. A., et al.
- Sommese, R. F., et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proceedings of the National Academy of Sciences of the United States of America. 110 (31), 12607-12612 (2013).
- Nag, S., et al. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nature Structural & Molecular Biology. 24 (6), 525-533 (2017).
- Kawana, M., Sarkar, S. S., Sutton, S., Ruppel, K. M., Spudich, J. A. Biophysical properties of human b-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Science Advances. 3 (2), 1-11 (2017).
- Girolami, F., et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circulation: Cardiovascular Genetics. 7 (6), 741-750 (2014).
- Debold, E. P., et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. American Journal of Physiology - Heart and Circulatory Physiology. 293 (1), 284-291 (2007).
- Barron, J. T.
- Duke, T. A. J.
- Vilfan, A., Duke, T.
- Huxley, A. F. Muscle structure and theories of contraction. Progress in Biophysics and Biophysical Chemistry. 7, 255-318 (1957).
- Huxley, H. E. Fifty years of muscle and the sliding filament hypothesis. European Journal of Biochemistry. 271 (8), 1403-1415 (2004).
- Kad, N. M., Kim, S., Warshaw, D. M., VanBuren, P., Baker, J. E. Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. Proceedings of the National Academy of Sciences of the United States of America. 102 (47), 16990-16995 (2005).
- Veigel, C., Molloy, J. E., Schmitz, S., Kendrick-Jones, J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nature Cell Biology. 5 (11), 980-986 (2003).
- Spudich, J. A.
- Simmons, R. M., Finer, J. T., Chu, S., Spudich, J. A. Quantitative measurements of force and displacement using an optical trap. Biophysical Journal. 70 (4), 1813-1822 (1996).
- Finer, J. T., Simmons, R. M., Spudich, J. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368 (6467), 113-119 (1994).
- Kron, S. J., Uyeda, T. Q. P., Warrick, H. M., Spudich, J. A. An approach to reconstituting motility of single myosin molecules. Journal of Cell Science. 98, 129-133 (1991).
- Molloy, J. E., Burns, J. E., Kendrick-Jones, B., Tregear, R. T., White, D. C. S. Movement and force produced by a single myosin head. Nature. 378 (6553), 209-212 (1995).
- Ruegg, C., et al. Molecular motors: Force and movement generated by single Myosin II molecules. Physiology. 17 (5), 213-218 (2002).
- Nayak, A., et al. Single-molecule analysis reveals that regulatory light chains fine-tune skeletal myosin II function. Journal of Biological Chemistry. 295 (20), 7046-7059 (2020).
- Dupuis, D. E., Guilford, W. H., Wu, J., Warshaw, D. M.
- Tyska, M. J., et al. Two heads of myosin are better than one for generating force and motion. Proceedings of the National Academy of Sciences of the United States of America. 96 (8), 4402-4407 (1999).
- Tyska, M. J., Warshaw, D. M.
- Finer, J. T., et al.
- Al Azzam, O., Trussell, C. L., Reinemann, D. N. Measuring force generation within reconstituted microtubule bundle assemblies using optical tweezers. Cytoskeleton. 78 (3), 111-125 (2021).
- Wagoner, J. A., Dill, K. A. Evolution of mechanical cooperativity among myosin II motors. Proceedings of the National Academy of Sciences of the United States of America. 118 (20), 2101871118 (2021).
- Walcott, S., Warshaw, D. M., Debold, E. P. Mechanical coupling between myosin molecules causes differences between ensemble and single-molecule measurements. Biophysical Journal. 103 (3), 501-510 (2012).
- Stewart, T. J., Murthy, V., Dugan, S. P., Baker, J. E. Velocity of myosin-based actin sliding depends on attachment and detachment kinetics and reaches a maximum when myosin-binding sites on actin saturate. Journal of Biological Chemistry. 297 (5), 101178 (2021).
- Hilbert, L., Cumarasamy, S., Zitouni, N. B., Mackey, M. C., Lauzon, A. M. The kinetics of mechanically coupled myosins exhibit group size-dependent regimes. Biophysical Journal. 105 (6), 1466-1474 (2013).
- Debold, E. P., Walcott, S., Woodward, M., Turner, M. A. Direct observation of phosphate inhibiting the Force-generating capacity of a miniensemble of myosin molecules. Biophysical Journal. 105 (10), 2374-2384 (2013).
- Kaya, M., Higuchi, H. Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science. 329 (5992), 686-689 (2010).
- Pertici, I., et al. A myosin II nanomachine mimicking the striated muscle. Nature Communications. 9 (1), 1-10 (2018).
- Cheng, Y. S., De Souza Leite, F., Rassier, D. E. The load dependence and the force-velocity relation in intact myosin filaments from skeletal and smooth muscles. American Journal of Physiology - Cell Physiology. 318 (1), 103-110 (2020).
- Stam, S., Alberts, J., Gardel, M. L., Munro, E. Isoforms confer characteristic force generation and mechanosensation by myosin II filaments. Biophysical Journal. 108 (8), 1997-2006 (2015).
- Rastogi, K., Puliyakodan, M. S., Pandey, V., Nath, S., Elangovan, R. Maximum limit to the number of myosin II motors participating in processive sliding of actin. Scientific Reports. 6, 1-11 (2016).
- Debold, E. P., Patlak, J. B., Warshaw, D. M. Slip sliding away: Load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophysical Journal. 89 (5), 34-36 (2005).
- Albert, P. J., Erdmann, T., Schwarz, U. S. Stochastic dynamics and mechanosensitivity of myosin II minifilaments. New Journal of Physics. 16, (2014).
- Erdmann, T., Schwarz, U. S. Stochastic force generation by small ensembles of myosin II motors. Physical Review Letters. 108 (18), 1-5 (2012).
- Guo, B., Guilford, W. H. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motility and the Cytoskeleton. 59 (4), 264-272 (2004).
- Miller-Jaster, K. N., Petrie Aronin, C. E., Guilford, W. H. A quantitative comparison of blocking agents in the in vitro motility assay. Cellular and Molecular Bioengineering. 5 (1), 44-51 (2012).
- Mansoon, A., Balaz, M., Albet-Torres, N., Rosengren, K. J. In vitro assays of molecular motors -- impact of motor-surface interactions. Frontiers in Bioscience. 13, 5732-5754 (2008).
- Persson, M., et al. Heavy meromyosin molecules extending more than 50 nm above adsorbing electronegative surfaces. Langmuir. 26 (12), 9927-9936 (2010).
- Kron, S. J., Spudich, J. A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proceedings of the National Academy of Sciences of the United States of America. 83 (17), 6272-6276 (1986).
- Yanagida, T., Nakase, M., Nishiyama, K., Oosawa, F. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature. 307 (5946), 58-60 (1984).
- Tsuda, Y., Yasutake, H., Ishijima, A., Yanagida, T. Torsional rigidity of single actin filaments and actin-actin bond breaking force under torsion measured directly by in vitro micromanipulation. Proceedings of the National Academy of Sciences of the United States of America. 93 (23), 12937-12942 (1996).
- Stewart, T. J., et al. Actin sliding velocities are influenced by the driving forces of actin-myosin binding. Cellular and Molecular Bioengineering. 6 (1), 26-37 (2013).
- Harada, Y., Sakurada, K., Aoki, T., Thomas, D. D., Yanagida, T. Mechanochemical coupling in actomyosin energy transduction by in vitro movement assay. Journal of Molecular Biology. 216 (1), 49-68 (1990).
- Fordyce, P. M., Valentine, M. T., Block, S. M. Advances in surface-based assays for single molecules. Single-Molecule Techniques: A Laboratory Manual. , 431-460 (2008).
- Ozeki, T., et al. Surface-bound casein modulates the adsorption and activity of kinesin on SiO2 surfaces. Biophysical Journal. 96 (8), 3305-3318 (2009).
- Neuman, K. C., Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nature Methods. 5 (6), 491-505 (2008).
- Neuman, K. C., Block, S. M.
- Thoresen, T., Lenz, M., Gardel, M. L. Thick filament length and isoform composition determine self-organized contractile units in actomyosin bundles. Biophysical Journal. 104 (3), 655-665 (2013).
- Matusovsky, O. S., et al. Millisecond conformational dynamics of skeletal Myosin II power stroke studied by high-speed atomic force microscopy. ACS Nano. 15 (2), 2229-2239 (2021).
- Reinemann, D. N., et al. Collective force regulation in anti-parallel microtubule gliding by dimeric Kif15 kinesin motors. Current Biology. 27 (18), 2810-2820 (2017).
- Cordova, J. C., et al. Bioconjugated core-shell microparticles for high-force optical trapping. Particle and Particle Systems Characterization. 35 (3), 1-8 (2018).
- Reinemann, D. N., Norris, S. R., Ohi, R., Lang, M. J. Processive Kinesin-14 HSET exhibits directional flexibility depending on motor traffic. Current Biology. 28 (14), 2356-2362 (2018).
- Forth, S., Hsia, K. C., Shimamoto, Y., Kapoor, T. M. Asymmetric friction of nonmotor MAPs can lead to their directional motion in active microtubule networks. Cell. 157 (2), 420-432 (2014).
- Shimamoto, Y., Kapoor, T. M. Analyzing the micromechanics of the cell division apparatus. Methods in Cell Biology. 145, 173-190 (2018).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Thoresen, T., Lenz, M., Gardel, M. L.
- Murrell, M., Thoresen, T., Gardel, M.
- Weirich, K. L., Stam, S., Munro, E., Gardel, M. L. Actin bundle architecture and mechanics regulate myosin II force generation. Biophysical Journal. 120 (10), 1957-1970 (2021).
