

Summary
Se presenta y discute la formación de haces de actomiosina in vitro y la medición de la generación de fuerza del conjunto de miosina utilizando pinzas ópticas.
Abstract
Las miosinas son proteínas motoras que hidrolizan ATP para avanzar a lo largo de las pistas de filamentos de actina (FA) y son esenciales en procesos celulares como la motilidad y la contracción muscular. Para comprender sus mecanismos de generación de fuerza, la miosina II se ha investigado tanto a nivel de molécula única (SM) como como equipos de motores in vitro utilizando métodos biofísicos como el atrapamiento óptico.
Estos estudios mostraron que el comportamiento generador de fuerza de miosina puede diferir mucho cuando se mueve desde el nivel de una sola molécula en una disposición de tres perlas a grupos de motores que trabajan juntos en una superficie rígida de talón o cubreobjetos en una disposición de deslizamiento. Sin embargo, estas construcciones de ensayo no permiten evaluar la dinámica de grupo de la miosina dentro de la jerarquía estructural viscoelástica como lo harían dentro de una célula. Hemos desarrollado un método que utiliza pinzas ópticas para investigar la mecánica de la generación de fuerza por conjuntos de miosina que interactúan con múltiples filamentos de actina.
Estos paquetes de actomiosina facilitan la investigación en un entorno jerárquico y compatible que captura la comunicación motora y la salida de fuerza del conjunto. La naturaleza personalizable del ensayo permite alterar las condiciones experimentales para comprender cómo las modificaciones al conjunto de miosina, el haz de filamentos de actina o el entorno circundante dan como resultado diferentes salidas de fuerza.
Introduction
Las proteínas motoras son esenciales para la vida, convirtiendo la energía química en trabajo mecánico 1,2,3. Los motores de miosina interactúan con los filamentos de actina dando pasos a lo largo de los filamentos similares a una pista, y la dinámica de las redes actina-miosina lleva a cabo la contracción muscular, la motilidad celular, el anillo contráctil durante la citocinesis y el movimiento de la carga dentro de la célula, entre otras tareas esenciales 3,4,5,6,7,8 . Dado que las miosinas tienen tantas funciones esenciales, la falla en la funcionalidad de la red miosina-actina puede conducir al desarrollo de enfermedades, como mutaciones en la cadena pesada de miosina que causan hipercontractilidad cardíaca en la miocardiopatía hipertrófica (MCH)9,10,11,12,13,14 . En la contracción muscular, los motores individuales de miosina cooperan entre sí trabajando como un conjunto para proporcionar la energía mecánica requerida que lleva a cabo el deslizamiento relativo de las FA 4,15,16,17,18. Los motores de miosina forman puentes cruzados entre los AF y utilizan cambios conformacionales debido a su ciclo mecanoquímico para moverse colectivamente hacia el extremo de púas de los filamentos alineados 17,18,19,20,21.
El desarrollo de ensayos cuantitativos de motilidad in vitro a nivel de SM utilizando técnicas como la captura óptica ha facilitado la recopilación de detalles sin precedentes sobre cómo funcionan los motores de miosina individuales, incluida la medición de la generación de fuerza SM y los tamaños de paso 22,23,24,25,26,27,28,29,30 . Finer et al. desarrollaron el ensayo de atrapamiento óptico de "tres perlas" o "mancuernas" para sondear la mecánica de generación de fuerza de motores de miosina II23,31. Como la miosina muscular II trabaja en equipos para contraer AF pero no es procesiva a nivel SM, la orientación del ensayo de captura óptica tuvo que reorganizarse a partir del enfoque clásico de perlas ligadas al motor32. Para formar el ensayo de mancuernas, se utilizaron dos trampas ópticas para sostener un AF sobre un motor de miosina unido a un cordón adherido a un cubreobjetos, y la salida de fuerza del motor único se midió a través de los movimientos del AF dentro de la trampa23.
Sin embargo, las fuerzas SM y el uso de una orientación de ensayo de un solo motor / filamento único no dan una imagen completa sobre la generación de fuerza a nivel de sistema, ya que muchas proteínas motoras, incluida la miosina II, no funcionan de forma aislada y, a menudo, no funcionan como una suma de sus partes 15,16,17,32,33,34,35,36 . Son necesarias estructuras más complejas que incluyan más de un motor interactuando con más de un filamento para comprender mejor la sinergia de las redes de filamentos de miosina y actina15,32. La orientación del ensayo con mancuernas se ha explotado para investigar la generación de fuerza de conjuntos pequeños al tener múltiples miosinas unidas a un cordón o usar un filamento grueso de miosina unido a una superficie y permitir que los motores interactúen con el AF suspendido 4,23,34,37,38,39,40.
Otros ensayos de conjunto pequeño incluyen un ensayo de deslizamiento de filamentos in vitro en el que los motores de miosina están recubiertos sobre una superficie de cubreobjetos, y se utiliza un cordón unido a un AF para sondear la fuerza generada por el equipo de motores 4,35,36,38,39,40,41,42,43 . En ambos casos, las miosinas están unidas a una superficie rígida (perla o cubreobjetos) y utilizan un AF. En estos casos, los motores no son capaces de moverse libremente o comunicarse entre sí, ni tener miosinas rígidamente unidas refleja el ambiente jerárquico y compatible en el que los motores trabajarían juntos en el sarcómero32. Estudios previos han sugerido que la miosina II puede detectar su entorno y adaptarse en consecuencia a las cambiantes condiciones de concentración viscoelástica o motora alterando características como la generación de fuerza y la relación de trabajo41,44,45. Por lo tanto, existe la necesidad de desarrollar un ensayo de captura óptica que fomente y capture la comunicación motora y el cumplimiento del sistema para pintar una imagen más realista de los fundamentos mecanicistas de la generación de fuerza del conjunto de miosina II.
Aquí, desarrollamos un método para acoplar la estructura jerárquica in vitro con la captura óptica formando haces de actomiosina o sándwiches que consisten en múltiples motores de miosina que interactúan entre dos filamentos de actina. Esta geometría de ensayo modular tiene la capacidad de sondear directamente cómo los factores moleculares y ambientales influyen en la generación de fuerza de miosina en conjunto. Además, la investigación de los mecanismos de generación de fuerza a través de estos conjuntos de actina-miosina tiene el potencial de ayudar a modelar y comprender cómo las tareas celulares a gran escala, como la contracción muscular, se propagan desde el nivel molecular 9,10,13.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Cubreobjetos de grabado
- Disolver 100 g de KOH en 300 ml de etanol al 100% en un vaso de precipitados de 1.000 ml. Revuelva con una barra de agitación hasta que la mayoría del KOH se haya disuelto.
PRECAUCIÓN: La solución concentrada de KOH puede causar quemaduras y daños a la ropa. Use guantes, protección para los ojos y una bata de laboratorio. - Coloque los cubreobjetos individualmente en los bastidores de limpieza de cubreobjetos.
NOTA: Los bastidores están diseñados con ranuras que sostienen cubreobjetos individuales separados para permitir el grabado y el enjuague en cada cara del cubreobjetos, orificios de drenaje en la parte inferior y están hechos de material que puede soportar las duras condiciones de grabado. Pueden ser hechos a medida o comprados comercialmente. - Prepare y etiquete tres vasos de precipitados de 1,000 ml: uno con 300 ml de etanol y dos vasos con 300 ml de agua de ósmosis inversa (RO).
NOTA: Aquí, el agua RO se obtuvo de un purificador de agua de laboratorio, pero también se puede comprar comercialmente si no hay un purificador local disponible. - Coloque cada uno de los cuatro vasos de precipitados en un sonicador de baño para desgasificar durante 5 minutos.
- Sumerge un estante de cubreobjetos en el vaso de precipitados de KOH y etanol y sonicate durante 5 min.
- Transfiera el estante de cubreobjetos del vaso de precipitados KOH/etanol al vaso de precipitados de etanol solo. Sumerja el bastidor hacia arriba y hacia abajo en el vaso de precipitados hasta que no haya cuentas.
NOTA: Tenga cuidado de no perturbar los cubreobjetos o dejar caer con fuerza la rejilla en el vaso de precipitados. Esto hará que los cubreobjetos salgan del estante o causen salpicaduras químicas. - Transfiera con cuidado el estante de cubreobjetos del vaso de precipitados de etanol a un vaso de precipitados de agua, sumergiéndolo hacia arriba y hacia abajo hasta que no haya cuentas.
- Sumerge el estante de cubreobjetos en el vaso de precipitados de agua que aún no se ha utilizado y sonica de nuevo durante 5 min.
- Use una botella para rociar el estante de cubreobjetos con agua hasta que se escurra suavemente fuera de los cubreobjetos. Repita con el etanol.
- Colocar las rejillas a secar en un horno a 90 °C durante 20 min. Almacene los bastidores de cubreobjetos grabados a temperatura ambiente en recipientes cerrados para evitar la contaminación antes de su uso.
2. Polimerización de filamentos de actina
- Hacer solución T
- En un tubo cónico de 50 ml, añadir 3,94 g de Tris-HCl y 0,147 g de CaCl2. Agregue agua RO para hacer un volumen total de 50 ml y mezcle bien.
NOTA: Las concentraciones finales de la solución T son 500 mM Tris-HCl y 20 mM CaCl2, respectivamente. - Etiquetar el tubo Solución T y conservarlo a 4 °C.
- En un tubo cónico de 50 ml, añadir 3,94 g de Tris-HCl y 0,147 g de CaCl2. Agregue agua RO para hacer un volumen total de 50 ml y mezcle bien.
- Hacer búfer TC
- Mezcle 40 ml de agua RO y 1,5 ml de solución T en un tubo cónico de 50 ml. Cambie el pH a 8.0 agregando pequeñas cantidades de KOH concentrado. Agregue agua para hacer 50 ml de la solución y verifique el pH. Ajuste el pH si es necesario.
NOTA: El tampón TC final contiene 5 mM de Tris-HCl y 0,2 mM de CaCl2 a pH 8. - Etiquetar el tubo TC y almacenarlo a 4 °C.
- Mezcle 40 ml de agua RO y 1,5 ml de solución T en un tubo cónico de 50 ml. Cambie el pH a 8.0 agregando pequeñas cantidades de KOH concentrado. Agregue agua para hacer 50 ml de la solución y verifique el pH. Ajuste el pH si es necesario.
- Hacer FC Buffer
- Agregue 85 ml de agua de ósmosis inversa, 10 ml de solución T, 3,73 g de KCl y 0,041 g de MgCl2 a un frasco tampón de 100 ml. Modifique el pH a 7.5 agregando pequeños volúmenes de KOH concentrado. Agregue agua para hacer un volumen final de 100 mL y verifique el pH.
NOTA: El tampón FC final contiene 500 mM de Tris-HCl, 500 mM de KCl, 2 mM de MgCl 2 y 2 mM de CaCl2 a pH 7,5. - Etiquetar el tubo FC y almacenarlo a 4 °C.
- Agregue 85 ml de agua de ósmosis inversa, 10 ml de solución T, 3,73 g de KCl y 0,041 g de MgCl2 a un frasco tampón de 100 ml. Modifique el pH a 7.5 agregando pequeños volúmenes de KOH concentrado. Agregue agua para hacer un volumen final de 100 mL y verifique el pH.
- Prepare el tampón de actina general (GAB).
- Mezclar 485 μL de tampón TC, 10 μL de ATP de 10 mM y 5 μL de TDT de 50 mM en un tubo de microcentrífuga.
NOTA: Las condiciones finales del búfer son 5 mM Tris-HCl, 0,2 mM CaCl 2, 0,5 mM DTT y0,2 mM ATP. - Etiquételo como GAB y guárdelo a 4 °C.
- Mezclar 485 μL de tampón TC, 10 μL de ATP de 10 mM y 5 μL de TDT de 50 mM en un tubo de microcentrífuga.
- Prepare el tampón de polimerización de actina (APB).
- Mezclar 455 μL de tampón FC, 25 μL de ATP de 100 mM y 20 μL de TDT de 50 mM en un tubo de microcentrífuga.
NOTA: Las condiciones finales del búfer son 50 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2, 2 mM CaCl2 2 mM TDT y 5 mM ATP. - Etiquetar el tubo como APB y almacenarlo a 4 °C.
- Mezclar 455 μL de tampón FC, 25 μL de ATP de 100 mM y 20 μL de TDT de 50 mM en un tubo de microcentrífuga.
- Reconstituir actina
- Reconstituir la actina del músculo esquelético del conejo añadiendo 100 μL de agua desionizada a un vial de 1 mg de actina liofilizada. Mezclar bien pipeteando suavemente hacia arriba y hacia abajo. Alícuota en muestras de 5 μL, congelar rápidamente y almacenar las alícuotas de actina de 10 mg/ml a -80 °C.
- Reconstituir la actina del músculo esquelético del conejo biotinilado añadiendo 20 μL de agua RO. Alícuota en muestras de 5 μL, congelar a presión y almacenar las alícuotas de actina biotiniladas de 1 mg/ml a -80 °C.
- Polimerización de actina no marcada con estabilización de faloidina rodamina
- Descongele un vial de 10 mg/ml de actina y manténgalo en hielo.
- Prepare un tampón de GAB fresco, agregue 100 μL de GAB a la alícuota de actina y mezcle pipeteando suavemente hacia arriba y hacia abajo. Incubar la solución en hielo durante 1 h.
- Preparar APB fresco durante la incubación. Después de la incubación, polimerizar la actina en filamentos añadiendo 11 μL de APB a la solución de actina. Mezclar bien pipeteando suavemente hacia arriba y hacia abajo. Colocar sobre hielo durante 20 min.
- Agregue 5 μL de faloidina marcada con rodamina a la solución de filamento de actina recién polimerizada. Dejar en hielo en la oscuridad durante 1 h.
- Guarde el vial de rodamina y actina envuelto en papel de aluminio en la oscuridad a 4 °C.
NOTA: Se sugiere utilizar estos filamentos durante un período máximo de 1 semana. La calidad del AF se puede confirmar cada día a través de una imagen rápida de una celda de flujo que contiene solo AF y la visualización de filamentos consistentes día a día.
- Polimerización de actina biotinilada con estabilización de faloidina Alexa Fluor 488
- Descongele un vial de 10 mg/ml de actina y 1 vial de 1 mg/ml de actina biotinilada y manténgalos en hielo.
- Haga un nuevo búfer GAB.
- Combinar los dos viales (paso 2.8.1) en una proporción 10:1 actina:actina biotinilada. Añadir 100 μL de GAB a la mezcla de actina y mezclar bien pipeteando suavemente hacia arriba y hacia abajo. Incubar en hielo durante 1 h.
- Hacer APB fresco durante la incubación.
- Después de la etapa de incubación, polimerizar la actina añadiendo 11 μL de APB a la solución de actina. Mezclar bien pipeteando hacia arriba y hacia abajo suavemente. Incubar en hielo durante 20 min.
- Agregue 5 μL de faloidina marcada con Alexa Fluor 488 e incube en hielo en la oscuridad durante 1 h.
- Conservar el vial de actina biotinilada envuelto en papel de aluminio en la oscuridad a 4 °C.
NOTA: Estos filamentos se pueden utilizar durante un periodo máximo de 1 semana.
3. Preparación de miosina y perlas
- Reconstituir la miosina II
- Gire brevemente (~5 s) la miosina esquelética liofilizada II para recogerla en el fondo del tubo usando una minicentrífuga estándar.
- Reconstituir la miosina a 10 mg/ml añadiendo 100 μL de 1 mM de TDT preparado en agua RO.
- Diluir la solución madre de miosina 10 veces añadiendo 10 μL de 10 mg/ml de miosina a 90 μL de 1 mM de DTT en agua RO. Hacer alícuotas de volumen pequeño (1-5 μL), congelarlas a presión y almacenar a -80 °C.
NOTA: La actividad de la miosina se puede confirmar mediante la realización de un ensayo de filamento deslizante estándar como se publicó anteriormente46,47. Vea la discusión para una breve descripción.
- Limpieza de perlas recubiertas de estreptavidina
- Diluir 20 μL de perlas de estreptavidina de 1 μm en 80 μL de agua RO. Lavar cuatro veces girando a 9.600 × g y reconstituyendo en 100 μL de agua RO.
- Sonicar durante 2 min a una amplitud del 40% y almacenar las perlas lavadas en un rotador a 4 °C.
4. Preparación de la celda de flujo
- Prepare una solución de poli-l-lisina (PLL) agregando 30 ml de etanol al 100% a un tubo de 50 ml y agregando 200 μL de poli-l-lisina al 0,1% p/v en agua y mezcle bien.
- Agregue un cubreobjetos grabados a la solución de PLL y déjela en remojo durante 15 minutos. Retire el cubreobjetos con unas pinzas, teniendo cuidado de tocar únicamente el borde del cubreobjetos a medida que se levanta del tubo (véase la figura 1A-C). Agarra los cubreobjetos por sus bordes con una mano enguantada.
- Seque el cubreobjetos con una línea aérea filtrada hasta que no quede etanol ni residuos en el cubreobjetos.
- Aplique dos piezas de cinta adhesiva de doble cara en el centro de un portaobjetos de microscopio, a 3-4 mm de distancia entre sí. Rasgue o corte el exceso de cinta que cuelga del borde de la diapositiva.
- Agregue el cubrecubierto con PLL en la parte superior de la cinta perpendicular al eje largo del portaobjetos del microscopio (formando una T) para formar un canal.
- Use un tubo pequeño para comprimir el cubreobjetos en la cinta y el portaobjetos del microscopio a fondo hasta que la cinta sea transparente (Figura 1A). Asegúrese de que no haya burbujas en la cinta, ya que esto puede causar fugas del canal de flujo.
NOTA: La celda de flujo puede contener un volumen de 10-15 μL.
5. Preparación del haz de actomiosina
- En tubos separados, diluir cada tipo de filamento de actina (marcado con rodamina y biotinilado 488) 600x mezclando 0,5 μL de la respectiva actina marcada con 300 μL de APB. Agregue 5 μL adicionales de faloidina correspondientemente etiquetada a cada tubo e incube en hielo en la oscuridad durante 15 minutos.
- A la solución de actina biotinilada, añadir un sistema de eliminación de oxígeno de 1 μL de beta-D-glucosa a 500 mg/ml, 1 μL de glucosa oxidasa a 25 mg/ml y 1 μL de catalasa a 500 unidades/ml. Añadir 1 μL de ATP de 100 mM y 1 μL de perlas de estreptavidina diluidas y limpias 100x. Revuelva suavemente con la punta de una pipeta. Coloque la suspensión en un rotador a 4 °C mientras se monta el resto del haz de actomiosina.
- Añadir 15 μL de la actina rodamina diluida a la célula de flujo PLL (Figura 1D). Absorba el exceso de solución a través de la celda de flujo, pero no permita que el canal de flujo se seque. Incubar durante 10 min en una cámara de humedad.
NOTA: Las cámaras de humedad se pueden hacer de cajas de punta de pipeta vacías con agua agregada a la parte inferior y la tapa cubierta con papel de aluminio para bloquear la luz. - Prepare una solución de caseína de 1 mg/ml en APB.
- Añadir 15 μL de 1 mg/ml de caseína para evitar la unión inespecífica de los componentes posteriores (Figura 1E). Incubar durante 5 min en una cámara de humedad.
- Añadir la concentración deseada de miosina a la suspensión de actina y perlas biotiniladas a partir del paso 5.2. Revuelva suavemente con la punta de la pipeta y, a continuación, agregue inmediatamente 15 μL de la suspensión del paso 5.2 + la concentración de miosina deseada a la celda de flujo (Figura 1F, G). Incubar durante 20 min. Selle los extremos abiertos de la celda de flujo con esmalte de uñas para evitar la evaporación durante los experimentos de captura óptica y de imágenes.
NOTA: Una concentración de solución de miosina de 1 μM produce un agrupamiento robusto y se puede utilizar como punto de partida para la personalización deseada del ensayo (ver Figura 2).
6. Mediciones de fuerza mediante trampa óptica (NT2 Nanotracker2)
NOTA: Si bien el siguiente protocolo es específicamente para el sistema NT2, este ensayo se puede usar con otros sistemas de captura óptica, incluidos los que están hechos a medida, que también tienen capacidades de fluorescencia. El flujo de trabajo general sigue siendo el mismo de enfocar la superficie de la diapositiva, realizar calibraciones de cuentas y adquirir datos mediante la búsqueda de haces de actina fluorescente. Para el sistema NT2, la figura suplementaria S1, la figura suplementaria S2, la figura suplementaria S3, la figura suplementaria S4, la figura suplementaria S5, la figura suplementaria S6 y la figura suplementaria S7 proporcionan detalles del sistema de captura óptica y la interfaz del software.
- Encienda la caja de control y el láser (Figura suplementaria S1).
- Inicie el software de la computadora de trampa óptica haciendo clic en el icono JPK Nanotracker en el escritorio.
- Activa el mando a distancia haciendo clic en el botón Logitech situado en el centro (figura suplementaria S2).
- Encienda el módulo de fluorescencia activando el interruptor de encendido/apagado (Figura suplementaria S3).
- Gire la torreta del cubo del filtro para obtener imágenes de campo claro (Figura suplementaria S4).
- Una vez que el sistema esté listo, encienda el láser usando el botón de encendido láser en la esquina inferior izquierda de la pantalla a 50 mW y deje que se estabilice durante 30 minutos (Figura suplementaria S5).
- Haga clic secuencialmente en los botones Iluminación, Cámara, Objetivo y Movimiento de escenario dentro del software para abrir esas ventanas para ver y manipular durante el experimento. Encienda la iluminación del microscopio haciendo clic en el botón de encendido/apagado y ajustándola a máxima potencia haciendo clic y arrastrando la barra hasta la derecha (Figura suplementaria S5).
- Abra el área de muestra y retire el portamuestras de la etapa del microscopio. Agregue la celda de flujo, asegúrela con los soportes de muestras de metal y asegúrese de que la diapositiva con el cubreobjetos esté en la parte inferior.
- Agregue 30 μL de agua RO al centro del objetivo inferior. No deje que la punta de la pipeta toque la lente. Vuelva a insertar la etapa de ejemplo.
NOTA: Como el sistema NT2 utiliza un objetivo de inmersión en agua como objetivo de captura, el medio de inmersión puede ser diferente dependiendo del objetivo de captura en la configuración del usuario. - Levante el objetivo inferior con las flechas de control en pantalla o L2 en el control remoto hasta que la cuenta de agua toque el cubreobjetos (Figura suplementaria S5).
- Baje el objetivo superior hasta que se alcance aproximadamente la mitad de la distancia a la celda de flujo utilizando las flechas en pantalla o R2 en el control remoto. Agregue 170 μL de agua RO a la parte superior de la celda de flujo directamente debajo del objetivo superior. Baja el objetivo superior hasta que rompa la tensión superficial del agua y forme un menisco.
- Mueva la plataforma del microscopio con la almohadilla de flecha del mando a distancia hasta alcanzar el borde de la cinta adyacente al canal de flujo. Cierre la puerta de muestra.
NOTA: Un "clic" al cerrar la puerta de muestra indica que el obturador láser ahora está abierto. Esta es una característica de seguridad que solo permite que el obturador se abra si la puerta está cerrada. - Usando la ventana Objetivo en la pantalla, enfoca el borde de la cinta levantando el objetivo inferior llamado Objetivo láser haciendo clic en la flecha superior usando los controles en pantalla. Haga lo mismo para el objetivo superior haciendo clic en la flecha inferior (Figura suplementaria S5).
NOTA: Las flechas dobles mueven el objetivo o la etapa más rápido. El borde de la cinta se utiliza para enfocar porque es un objeto grande y fácil de encontrar que está cerca de la superficie del cubreobjetos. Las burbujas de aire dentro de la cinta son otra opción. Sin embargo, esto no es necesario si el usuario tiene una rutina automatizada para encontrar el enfoque de superficie o un método interno preferido. - Una vez que la cinta esté enfocada, cierre parcialmente el iris en la parte superior de la trampa óptica. Coloca el objetivo superior hacia abajo hasta que la forma poligonal del iris sea visible. Enfoca esos bordes, vuelve a abrir el iris y luego acopla los objetivos haciendo clic en el icono del candado (Figura suplementaria S5).
- Encuentra una cuenta flotante y trampérala haciendo clic en el botón Trap Shutter , que abrirá el obturador y permitirá que el láser de captura golpee la muestra. Haga clic en el cursor Trap en la pantalla y arrástrelo para mover la ubicación del láser de captura. Una vez atrapado, calibre el talón para correlacionar las mediciones de voltaje con la fuerza y el desplazamiento.
- Haga clic en el botón Calibración . Ajuste la rutina de calibración basada en el análisis de espectros de potencia y ajuste la frecuencia de esquina dentro del software para las direcciones X, Y y Z (Figura suplementaria S6).
- Haga clic en Configuración. Escriba el diámetro de la cuenta (1,000 nm) y escriba la temperatura de la etapa que se encuentra en la parte inferior izquierda de la ventana del software. (véase la figura suplementaria S6).
- Haga clic en Trampa 1. Haga clic en X Signal. Haga clic en Ejecutar para realizar el ajuste de frecuencia de esquina. Haga clic y arrastre dentro de la ventana para optimizar el ajuste de la función. Haga clic en Usarlo para conocer los valores de sensibilidad y rigidez. Haga clic en Aceptar valores. Repita para las señales Y y Z. Cierre la ventana. (véase la figura suplementaria S6).
NOTA: Las rutinas de calibración de cuentas en otros sistemas de captura óptica o sistemas personalizados que han sido probados sólidamente por el usuario, como el método de equipartición o el método de fuerza de arrastre, también son aceptables57,58. - Encuentre un paquete de actomiosina buscando cuentas unidas a AF en la superficie del cubreobjetos.
- Cuando se detecta una cuenta no abarrotada por otras cuentas flotantes, observe los AF a su alrededor mediante imágenes de fluorescencia para verificar la presencia de un paquete.
- Verifique que un paquete esté presente buscando ambos AF fluorescentes colocalizados. Encienda la fuente de luz blanca y use el cubo de filtro apropiado para obtener imágenes de cada filamento de actina girando la torreta (cubos de filtro de excitación de 488 nm y 532 nm para Alexa Fluor 488 y excitación de rodamina, respectivamente). Véase la figura suplementaria S4.
NOTA: Un experimento de control para verificar la intensidad de fluorescencia de AF individuales puede ser útil para identificar haces que están compuestos por un solo filamento marcado con rodamina 488 y simples, o aplicable a cualquier conjunto de fluoróforos que el usuario elija usar. - Una vez verificado, atrape la cuenta unida al filamento superior del paquete haciendo clic en el botón Obturador de trampa .
- Utilice los controles en pantalla para registrar los datos haciendo clic en el botón Osciloscopio (Figura suplementaria S7). Para visualizar las mediciones sin registrar los datos, haga clic en Inicio. Para guardar todos los datos, haga clic en Autoguardar. Para registrar las mediciones, haga clic en Iniciar registro. Elija qué datos deben visualizarse en tiempo real (posición, fuerza, dirección x, dirección y) eligiendo en el menú desplegable señal X o señal Y. Recuerde que xdirection es de izquierda a derecha, y la dirección y está arriba y abajo en la pantalla. Véase la figura suplementaria S7.
NOTA: Los datos se guardarán como archivos .out e incluyen tiempo, voltaje, desplazamiento y fuerza para cada dirección. Estos archivos se pueden exportar a otro software para su visualización y análisis.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Las células de flujo que contienen los sistemas de haz de actomiosina son de un diseño estándar, que consiste en un portaobjetos de microscopio y un cubreobjetos grabados separados por un canal hecho de cinta adhesiva de doble cara (Figura 1). Luego, el ensayo se construye desde el cubreobjetos utilizando introducciones escalonadas como se describe en el protocolo. El ensayo final consiste en filamentos de actina marcados con rodamina plantilla; la concentración de miosina deseada (se utilizó 1 μM para los resultados representativos en la Figura 2 y la Figura 3); filamentos de actina biotinilados, marcados con Alexa Fluor 488; 1 μm de perlas de estreptavidina; el sistema de eliminación de oxígeno; ATP; y búfer APB. Se formarán múltiples haces por celda de flujo, y las concentraciones de actina descritas anteriormente proporcionan un espacio adecuado entre los haces para garantizar que no haya interacciones no deseadas. Esto también facilita la obtención de múltiples mediciones de fuerza por celda de flujo para aumentar la eficiencia de adquisición de datos. Los perfiles de fuerza deben ser reproducibles dentro de una celda de flujo y de celda de flujo a celda de flujo.
Si bien el protocolo anterior está orientado hacia el uso de una configuración comercial de captura óptica, la celda de flujo y el ensayo presentados aquí podrían utilizarse fácilmente para un instrumento comercial diferente o una configuración de captura óptica personalizada junto con un microscopio o una etapa de microscopio y que posean capacidades de imágenes de fluorescencia. Una vez que todas las adiciones de celdas de flujo se completan de acuerdo con el protocolo anterior, los haces de actomiosina en el portaobjetos (Figura 1) están listos para la medición inmediata. La celda de flujo se agrega a la etapa de microscopio de trampa óptica, se adquieren múltiples mediciones de calibración de perlas y se identifican haces a través de la colocalización de fluorescencia de los filamentos del haz. Una cuenta atada a un haz queda atrapada, y comienza el desplazamiento y la medición de la fuerza correspondiente. El usuario puede observar la adquisición de datos en tiempo real en el monitor de la computadora. Dependiendo de la concentración de miosina utilizada en la celda de flujo, el haz podría comenzar a exhibir un movimiento sustancial de inmediato, o puede tomar 30 s-1 min para ver efectivamente un aumento en el desplazamiento / fuerza.
Una traza de fuerza representativa se muestra en la Figura 3A donde los motores de miosina exhiben una rampa constante en fuerza seguida de una meseta. Es típico ver este tipo de rastros desarrollarse durante 2-5 min. Sin embargo, también es posible medir haces de actomiosina que no generan ninguna fuerza neta (Figura 3B). Estas trazas aparecen como ruido de referencia o no presentan un aumento neto sustancial de la fuerza durante 90 s. Esto es probablemente debido a una baja concentración local del motor que no permite el deslizamiento productivo, o el haz está en una orientación paralela desfavorable donde los extremos más y menos de los filamentos están alineados.
Como el contenido de la celda de flujo puede ser susceptible a la degradación de la iluminación incidente y el láser de captura, el calentamiento local en el portaobjetos con el tiempo y la generación de especies radicales de oxígeno, se recomienda encarecidamente no usar la misma celda de flujo durante más de 1 h. Para obtener la máxima eficiencia, se sugiere tener otro ensayo incubando mientras se adquieren datos. El rastreo de desplazamiento / fuerza se puede exportar desde el software de captura óptica a Excel, Matlab, Igor u otros programas de gestión de datos para su posterior filtrado y análisis. Los datos que pueden extraerse de tales experimentos de conjunto/haz de captura óptica incluyen diferentes tipos de perfiles de generación de fuerza (línea de base, rampa/meseta) bajo diferentes condiciones de ensayo, velocidad de generación de fuerza, generación de fuerza máxima, cinética de conjunto y comportamiento de paso a través de tamaños de paso y tiempos de permanencia entre pasos o equipos de pasos, así como relación de trabajo. El usuario también puede alterar las condiciones del ensayo para comparar cómo la adición de diferentes tipos de motores de miosina, la adición de proteínas de unión a actina o el cambio de las condiciones de amortiguación influyen en estas características de generación de fuerza de conjunto.
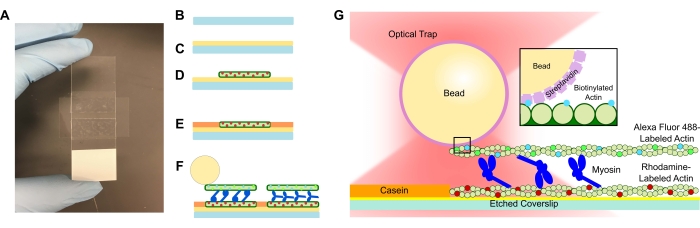
Figura 1: Esquema del ensayo. (A-C) Los cubreobjetos grabados están recubiertos de poli-L-lisina y se utilizan para formar la celda de flujo mediante el uso de cinta de doble cara y un portaobjetos de microscopio. Las introducciones cronometradas y los pasos de incubación descritos en el protocolo dan como resultado actina estabilizada con faloidina marcada con rodamina como molde o filamento inferior (D), seguida de bloqueo de caseína para evitar la unión no específica (E), y (F) actina biotinilada estabilizada con faloidina Alexa Fluor 488 como filamento de carga o superior, y equipos de miosina II que separan los filamentos y generan fuerza cuando se introduce ATP. La geometría de los motores y la naturaleza de la reticulación dentro del haz podrían variar en diferentes condiciones, como la concentración de sal59. Estudios previos han demostrado que el dominio de la cola de miosina tiene la capacidad de interactuar con los filamentos de actina y ralentizar la motilidad del conjunto46. Sin embargo, las cabezas de miosina en experimentos con meromiosina pesada demuestran la unión de cada cabeza a filamentos de actina adyacentes60. (G) Las perlas de estreptavidina se utilizan como mango óptico para la trampa y se unen únicamente al filamento de actina biotinilada de carga, lo que ayuda a validar que se forman haces adecuados en el portaobjetos. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Haces fluorescentes de actomiosina. Cuatro encuentros diferentes de filamentos de actina y haces dentro del ensayo de haz presentado en la Figura 1. El filamento de actina biotinilada de carga superior con el canal de faloidina Alexa Fluor 488 se muestra a la izquierda, y el filamento de actina de plantilla inferior con el canal de faloidina rodamina está a la derecha. En la parte inferior, se muestra la misma figura con líneas de colores superpuestas para ayudar a guiar el ojo. (A) Un filamento de actina superior se encuentra cerca de un filamento de actina inferior, pero tiene una superposición incompleta. Esto no se usaría para experimentos de paquete. (B) Los filamentos de actina superior e inferior están colocalizados, y la intensidad de cada filamento confirma que son filamentos individuales dentro del haz. Este sería un buen candidato para experimentos de paquete. (C) Un gran haz de filamentos de rodamina autoensamblados se encuentra en la parte inferior. Si bien hay un filamento de actina superior correspondiente que está colocalizado, hay demasiados filamentos inferiores presentes; por lo tanto, no se usaría para experimentos de paquete. Este es también un ejemplo de cómo cuando se agrupan múltiples filamentos de actina del mismo tipo, la intensidad de fluorescencia aumenta. El usuario puede utilizar esto como un indicador para juzgar filamentos individuales frente a paquetes del mismo tipo de filamento. (D) Un filamento inferior está presente sin el filamento superior correspondiente, lo que también confirma que no hay sangrado. Esto no se usaría para experimentos de paquete. Notamos que la intensidad de los filamentos en el canal Alexa Fluor 488 es baja y creemos que se debe al conjunto de filtros que se está utilizando (Filter Set 09 de Zeiss). El conjunto de filtros utilizado para el canal de rodamina es el conjunto de filtros 43 de Zeiss. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: Generación de fuerza de conjunto de miosina II. Trazas representativas de motores de miosina esquelética II que generan fuerza dentro de la jerarquía estructural de actina in vitro construida. Los motores de miosina están trabajando juntos para generar fuerza colectiva y productivamente hasta que se alcanza una meseta y se mantiene la fuerza (A) o experimentan antagonización cerca de la línea de base (B). Haga clic aquí para ver una versión más grande de esta figura.
Figura suplementaria S1: Trampa óptica Bruker/JPK Nanotracker2. (A) Monitor de computadora. (B) Teclado de computadora. (C) Torre de computadoras. (D) Caja del controlador. (E) Fuente de alimentación láser. (F) Caja óptica de trampa óptica. (G) Microscopio invertido. (H) Puerta a la etapa de microscopio. (I) Control deslizante polarizador para cambiar entre imágenes de contraste de campo claro e interferencia diferencial. Haga clic aquí para descargar este archivo.
Figura suplementaria S2: Control remoto para trampa óptica. (A) Teclado para colocar la platina motorizada. (B-C) Ajuste la posición de la trampa. (D) A, X y B encienden y apagan el obturador principal, el obturador de la trampa 1 y el obturador de la trampa 2, respectivamente. (E) El botón Logitech se utiliza para activar el mando. (F) Los botones arriba y abajo que se utilizan para colocar el objetivo de captura. (G) Los botones arriba y abajo que se utilizan para posicionar el objetivo de detección. Tenga en cuenta que el control remoto no es necesario, y todas estas manipulaciones se pueden realizar en el software. Sin embargo, es conveniente poder controlar los objetivos y la posición del escenario mientras se observa el entorno de la etapa del microscopio. Haga clic aquí para descargar este archivo.
Figura suplementaria S3: Módulo de fluorescencia para trampa óptica. La fuente de luz blanca de fluorescencia 89North PhotoFluor está acoplada a la parte posterior del microscopio invertido. Se enciende y apaga con un interruptor de palanca (flecha). Haga clic aquí para descargar este archivo.
Figura suplementaria S4: Torreta del cubo del filtro de fluorescencia. La torreta (flecha) se puede girar para usar el cubo de filtro necesario para obtener imágenes en tintes DIC, rodamina o Alexa Fluor 488. Tenga en cuenta que los cubos de filtro se pueden cambiar para personalizar la configuración para usar diferentes fluoróforos. Haga clic aquí para descargar este archivo.
Figura suplementaria S5: Software Nanotracker2. (A) Botón de encendido láser y control. (B) Ventana de posicionamiento objetivo. Las flechas direccionales se utilizan para mover los objetivos de detección (arriba) y de captura (abajo). Las flechas dobles mueven los objetivos a una velocidad mayor. El botón azul y rojo en la parte inferior izquierda desacopla los objetivos y los retrae a su posición original. Esto es necesario para cuando se toman muestras dentro y fuera de la etapa de microscopio. El tercer botón de la izquierda con los objetivos y el icono del candado "acopla" los objetivos para que cuando ambos estén enfocados y logren la iluminación de Kohler, el usuario pueda mover los objetivos de captura y detección hacia arriba y hacia abajo en el eje z. (C) Ventana de posicionamiento de la muestra utilizada para mover la etapa del microscopio en los ejes x e y. Las flechas dobles mueven el escenario a una velocidad más alta. Esta ventana se activa haciendo clic en el icono de flecha arriba/abajo e izquierda/derecha en el menú superior. (D) Ventana de visualización de la cámara. El icono de llave inglesa se puede utilizar para establecer condiciones de imagen personalizadas. Esta ventana se activa haciendo clic en el icono de la cámara en el menú superior. (E) Ventana de iluminación del microscopio. Esta ventana se activa haciendo clic en el icono de la bombilla en el menú superior. Haga clic aquí para descargar este archivo.
Figura suplementaria S6: Ventana de calibración. (A) Esta ventana se utiliza para la calibración de cuentas y se activa haciendo clic en el icono Cal en el menú superior. Para calibrar un talón, se logra un mejor ajuste de la frecuencia de esquina en las señales x, y y z. (B) Para cada señal, elija el botón de señal apropiado en la parte superior izquierda. (C) Haga clic en ejecutar y optimice el ajuste haciendo clic y arrastrando dentro de la ventana verde (D). (E) Una vez satisfecho con el ajuste, haga clic en Úselo para sensibilidad y rigidez. Esto permitirá registrar el desplazamiento en nanómetros y la fuerza en piconewtons. (F) Luego, haga clic en Aceptar valores en la parte inferior izquierda. Repita para las direcciones y y z. Haga clic aquí para descargar este archivo.
Figura suplementaria S7: Ventana de adquisición de datos. Esta ventana se utiliza para adquirir datos de posición y fuerza y permite al usuario ver las mediciones en tiempo real. (A) Esta ventana se activa haciendo clic en el icono x,t en el menú superior. (B) El usuario puede cambiar entre la visualización de las señales x e y. (C) Haga clic en Inicio para comenzar a visualizar los datos. Haga clic en Autoguardar para guardar los datos. Haga clic en Iniciar grabación para comenzar a grabar y guardar datos. Haga clic aquí para descargar este archivo.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Se realizó un estudio in vitro utilizando pinzas ópticas combinadas con imágenes de fluorescencia para investigar la dinámica de los conjuntos de miosina que interactúan con los filamentos de actina. Los haces de actina-miosina-actina se ensamblaron utilizando miosina muscular II, actina rodamina en la parte inferior del haz y en la superficie del cubreobjetos, y filamentos de actina biotinilados marcados con 488 en la parte superior del paquete. La proteína de actina del músculo del conejo se polimerizó y estabilizó utilizando tampones de actina generales (GAB) y tampones de polimerización de actina (APB). GAB y APB deben prepararse todos los días en el laboratorio utilizando ATP, tampón FC y tampón TC. La miosina muscular II se utilizó para formar los sándwiches de actina-miosina-actina. La faloidina se utilizó para la tinción fluorescente de los filamentos de actina, así como para la estabilización in vitro.
La actividad de la miosina se puede confirmar mediante la realización de un ensayo de filamento deslizante estándar como se publicó anteriormente46,47. La miosina II y sus subfragmentos pueden unirse a la superficie del cubreobjetos en una variedad de orientaciones, y la presencia del dominio de la cola puede ralentizar el deslizamiento del filamento en comparación con los ensayos con meromiosina pesada46,48,49. Sin embargo, todavía se puede observar el deslizamiento y el movimiento de la superficie. Una demostración más evidente de la actividad de la miosina es la rotura activa del filamento de actina que se puede observar donde los filamentos de actina más largos se rompen en fragmentos más pequeños que luego se deslizan en múltiples direcciones. Esto ocurre debido a la alta concentración de motores activos en la superficie, ha sido observado por múltiples laboratorios, y no ocurre sin motores de miosina activos presentes 42,50,51,52,53,54. Además, el ensayo de haz presentado aquí ayuda a aliviar los problemas de motilidad que se han asociado principalmente con el ensayo de filamento deslizante, como la variedad de orientaciones de unión del motor en un cubreobjetos de vidrio, porque el ensayo del haz implica el bloqueo de caseína de la superficie del vidrio para que los motores se unan dentro del haz 47,55,56.
El primer paso es agregar filamentos de actina de rodamina como filamento inferior o plantilla a un cubrecubierto recubierto de poli-L-lisina en una celda de flujo. La poli-L-lisina se utiliza para promover la unión a la actina ya que la polilisina está cargada positivamente, mientras que la actina tiene cargas negativas y se ha utilizado en preparaciones previas de ensayos citoesqueléticos in vitro 61,62,63. Antes de la formación del haz, se agregaron diferentes diluciones de actina a una celda de flujo para optimizar la concentración de actina. En este caso, 600x del stock fue la dilución óptima que produjo un número suficiente de filamentos de plantilla para la formación de paquetes, pero con un espaciado adecuado para que los paquetes fueran individualizados. La dilución se llevó a cabo utilizando el tampón APB. La adición de actina rodamina fue seguida por una capa de caseína para bloquear la superficie y evitar la unión no específica. La celda de flujo se incubó durante 30 minutos y se lavó después de la incubación con tampón para eliminar cualquier filamento de actina no unido. Finalmente, se agregó una combinación de miosina, 488/biotina actina y perlas recubiertas de estreptavidina a la célula de flujo para facilitar la formación del haz de actina-miosina. La concentración del cordón debe ser tal que haya suficientes para unir los haces ligados a la superficie y suficientes en suspensión para facilitar la calibración. Sin embargo, una concentración de perlas demasiado alta puede causar dificultades durante los experimentos de captura debido a que las perlas vecinas caen en la trampa láser e interrumpen la medición. Los motores de miosina se agregan a la combinación justo antes de inyectarla en el portaobjetos para que los motores de miosina no se agreguen preventivamente con la carga o el filamento de actina biotinilada superior y, por lo tanto, unirán la rodamina inferior para agrupar filamentos de actina biotinilados.
El sistema de captura óptica NT2 es una trampa óptica comercial con modalidades combinadas de campo claro, contraste de interferencia diferencial (DIC) e imágenes de epifluorescencia. Se combina con un microscopio invertido Zeiss AxioObserver 3 con objetivos de captura y detección de inmersión en agua 100x/NA 1.46 y 63x/NA 1.0. El sistema está equipado con la capacidad de captura de clic y arrastre de una trampa láser y se puede utilizar al obtener imágenes en cualquiera de las modalidades enumeradas anteriormente. Los haces formados se detectan y confirman mediante el uso de imágenes de fluorescencia. Tener una fuente de luz blanca con cubos de filtro apropiados (GFP / FITC y TRITC / CY3) permite un cambio rápido entre imágenes de filamentos. Los AF colocalizados se verificaron visualizando los AF en las diferentes longitudes de onda de excitación antes de tomar cada medición de fuerza utilizando pinzas ópticas. Como los filamentos pueden fotoblanquearse rápidamente incluso con un reactivo eliminador de oxígeno, se sugiere que los investigadores optimicen los parámetros de visualización, como la intensidad y el tiempo de exposición, antes de realizar los experimentos del paquete.
La captura óptica se empleó para tomar las mediciones de fuerza, utilizando las perlas de estreptavidina en presencia de ATP para unir el filamento de actina de carga biotinilada y activar la generación de fuerza de miosina como transductor de fuerza. Los datos de desplazamiento y fuerza versus tiempo obtenidos por captura óptica se extrajeron del software de captura para su análisis. Sin embargo, el software de captura comercial también proporciona rutinas de análisis que se pueden utilizar, o algoritmos personalizados en otros programas pueden ser programados por el usuario para visualizar y analizar los datos de captura. En los sistemas de captura óptica personalizados, el usuario puede tener láseres de excitación en lugar de una fuente de luz blanca con filtros, que también son aceptables para usar. Además, los tintes fluorescentes se pueden cambiar para adaptarse al equipo existente que un usuario puede tener si los espectros de emisión no se superponen y causan sangrado.
Observamos que el ensayo presentado es un ensayo de referencia que puede ser personalizado por el usuario dependiendo de su pregunta de investigación dentro del ámbito de la mecánica de conjuntos de actomiosina. El flujo de trabajo general también se puede aplicar a otros sistemas de conjuntos citoesqueléticos in vitro que pueden ser de interés, como los ensayos de haz de microtúbulos que forman modelos mínimos de huso mitótico 32,61,63,64,65,66. Las modificaciones podrían incluir, entre otras, cambiar las etiquetas de fluoróforos que se adaptan a la configuración existente del usuario; alteración de la concentración, construcción o isotipo de miosina; y la titulación de las condiciones de amortiguación, entre otros aspectos.
Los desafíos potenciales son posibles al realizar este ensayo. Al formar los haces de actina-miosina, la concentración de miosina dentro de los haces de actina puede no ser homogénea a través del portaobjetos. Para acomodar esto, se medirán múltiples paquetes en toda la diapositiva para garantizar que la distribución del motor y los perfiles de generación de fuerza se muestreen correctamente. También es difícil conocer la orientación del paquete si esto es necesario para la interpretación de los datos de fuerza. Por lo tanto, se deben realizar múltiples ensayos para cada paquete. También se podría incorporar el etiquetado del extremo del filamento de actina a través de gelsolin fluorescente o perlas recubiertas de gelsolin de un tamaño más pequeño que el mango de captura óptica. Las imágenes de fluorescencia también se pueden usar para observar las fuerzas de los componentes x e y para deducir la orientación del haz. Además, como el estado de agregación de miosina está altamente influenciado por la fuerza iónica del tampón con formación de filamentos gruesos que ocurren tras la dilución rápida de KCl, la concentración de sales tampón debe monitorearse adecuadamente67,68.
Estudios previos que utilizaron otros métodos in vitro, como los ensayos de deslizamiento, fueron útiles para identificar el papel de los dominios de miosina y estudiar la configuración y las interacciones entre la miosina y otras proteínas de unión a actina. Sin embargo, estos métodos tienen la desventaja de que la unión de miosina a una superficie rígida limitará el potencial de coordinación entre los motores de miosina y, por lo tanto, la retroalimentación de mecanodetección que se produce para determinar si el conjunto motor está en un modo de relación de trabajo alta o baja 33,35,41,69. Además, la captura óptica con redes motoras de miosina única no da una comprensión clara de cómo los motores de miosina interactúan entre sí y con los filamentos de actina. El protocolo desarrollado aquí permite la investigación de la dinámica del conjunto motor de miosina dentro de una red de actina jerárquica compatible. También es personalizable en términos de características del conjunto motor-filamento, como concentración, isoforma y entorno amortiguador, entre otros aspectos, para permitir una investigación sistemática. El protocolo presentado es una plataforma para futuros estudios de redes de actomiosina más complejas y mantiene la precisión de las mediciones de desplazamiento y generación de fuerza facilitadas por la captura óptica que tradicionalmente se ha utilizado para estudios de moléculas individuales.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen conflictos de intereses que declarar.
Acknowledgments
Este trabajo es apoyado en parte por la Beca de Investigación del Consejo de Estudiantes Graduados de la Universidad de Mississippi (OA), la Universidad de Mississippi Sally McDonnell-Barksdale Honors College (JCW, JER), el Consorcio de Subvenciones Espaciales de Mississippi bajo el número de subvención NNX15AH78H (JCW, DNR) y la Asociación Americana del Corazón bajo el número de subvención 848586 (DNR).
Materials
| Name | Company | Catalog Number | Comments |
| Actin protein (biotin): skeletal muscle | Cytoskeleton | AB07-A | Biotinylated actin protein |
| Actin protein, rabbit skeletal muscle | Cytoskeleton | AKL99-A | Actin protein |
| Alexa Fluor 488 Phalloidin | Invitrogen | A12379 | Actin stabilizer and Alexa Fluor 488 stain |
| ATP | Fisher scientific | BP413-25 | Required for actin assembly and myosin motility |
| Beta-D-glucose | Fisher scientific | MP218069110 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Blotting Grade Blocker (casein) | Biorad | 1706404 | Used to block surface from non-specific binding |
| CaCl2 | Fisher scientific | C79500 | Calcium chloride, provides the necessary control over the dynamics of actin myosin network |
| Catalase | Fisher scientific | ICN10040280 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Coverslips | Fisher scientific | 12544C | Used to make flow cells |
| DTT | Fisher scientific | AC327190010 | Used for buffer preparation |
| Ethanol | Fisher scientific | A4094 | Regent used for cleaning coverslips |
| Glucose oxidase | Fisher scientific | 34-538-610KU | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| KCl | Fisher scientific | P217-500 | Used for buffer preparation |
| KOH | Fisher scientific | P250-1 | Used to etch coverslips and adjust buffer pH |
| MgCl2 | Fisher scientific | M33-500 | Used for buffer preparation |
| Microscope slides | Fisher scientific | 12-544-2 | Used to make flow cells |
| Myosin II protein: rabbit skeletal muscle | Cytoskeleton | MY02 | Full length myosin motor protein isolated from rabbit skeletal muscle |
| Nanotracker2 | Bruker/JPK | NT2 | Optical trapping instrument |
| Poly-l-lysine | Sigma-Aldrich | P8920 | Facilities adhesion of actin filaments onto glass surface of the coverslip |
| Rhodamine Phalloidin | Cytoskeleton | PHDR1 | Actin stabilizer and rhodamine fluorescent stain |
| Streptavidin beads, 1 μm | Spherotech | SVP-10-5 | Optical trapping handle |
| Tris-HCl | Fisher scientific | PR H5121 | Used for buffer preparation |
References
- Goldstein, L. S. Kinesin molecular motors: transport pathways, receptors, and human disease. Proceedings of the National Academy of Sciences of the United States of America. 98 (13), 6999-7003 (2001).
- Lee Sweeney, H., Holzbaur, E. L. F.
- O'Connell, C. B., Tyska, M. J., Mooseker, M. S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochimica et Biophysica Acta - Molecular Cell Research. 1773 (5), 615-630 (2007).
- Kaya, M., Tani, Y., Washio, T., Hisada, T., Higuchi, H. Coordinated force generation of skeletal myosins in myofilaments through motor coupling. Nature Communications. 8, 1-13 (2017).
- Akhshi, T. K., Wernike, D., Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 71 (1), 1-23 (2014).
- Brawley, C. M., Rock, R. S. Unconventional myosin traffic in cells reveals a selective actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 106 (24), 9685-9690 (2009).
- Hartman, M. A., Spudich, J. A. The myosin superfamily at a glance. Journal of Cell Science. 125 (7), 1627-1632 (2012).
- Spudich, J. A., et al.
- Sommese, R. F., et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proceedings of the National Academy of Sciences of the United States of America. 110 (31), 12607-12612 (2013).
- Nag, S., et al. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nature Structural & Molecular Biology. 24 (6), 525-533 (2017).
- Kawana, M., Sarkar, S. S., Sutton, S., Ruppel, K. M., Spudich, J. A. Biophysical properties of human b-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Science Advances. 3 (2), 1-11 (2017).
- Girolami, F., et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circulation: Cardiovascular Genetics. 7 (6), 741-750 (2014).
- Debold, E. P., et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. American Journal of Physiology - Heart and Circulatory Physiology. 293 (1), 284-291 (2007).
- Barron, J. T.
- Duke, T. A. J.
- Vilfan, A., Duke, T.
- Huxley, A. F. Muscle structure and theories of contraction. Progress in Biophysics and Biophysical Chemistry. 7, 255-318 (1957).
- Huxley, H. E. Fifty years of muscle and the sliding filament hypothesis. European Journal of Biochemistry. 271 (8), 1403-1415 (2004).
- Kad, N. M., Kim, S., Warshaw, D. M., VanBuren, P., Baker, J. E. Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. Proceedings of the National Academy of Sciences of the United States of America. 102 (47), 16990-16995 (2005).
- Veigel, C., Molloy, J. E., Schmitz, S., Kendrick-Jones, J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nature Cell Biology. 5 (11), 980-986 (2003).
- Spudich, J. A.
- Simmons, R. M., Finer, J. T., Chu, S., Spudich, J. A. Quantitative measurements of force and displacement using an optical trap. Biophysical Journal. 70 (4), 1813-1822 (1996).
- Finer, J. T., Simmons, R. M., Spudich, J. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368 (6467), 113-119 (1994).
- Kron, S. J., Uyeda, T. Q. P., Warrick, H. M., Spudich, J. A. An approach to reconstituting motility of single myosin molecules. Journal of Cell Science. 98, 129-133 (1991).
- Molloy, J. E., Burns, J. E., Kendrick-Jones, B., Tregear, R. T., White, D. C. S. Movement and force produced by a single myosin head. Nature. 378 (6553), 209-212 (1995).
- Ruegg, C., et al. Molecular motors: Force and movement generated by single Myosin II molecules. Physiology. 17 (5), 213-218 (2002).
- Nayak, A., et al. Single-molecule analysis reveals that regulatory light chains fine-tune skeletal myosin II function. Journal of Biological Chemistry. 295 (20), 7046-7059 (2020).
- Dupuis, D. E., Guilford, W. H., Wu, J., Warshaw, D. M.
- Tyska, M. J., et al. Two heads of myosin are better than one for generating force and motion. Proceedings of the National Academy of Sciences of the United States of America. 96 (8), 4402-4407 (1999).
- Tyska, M. J., Warshaw, D. M.
- Finer, J. T., et al.
- Al Azzam, O., Trussell, C. L., Reinemann, D. N. Measuring force generation within reconstituted microtubule bundle assemblies using optical tweezers. Cytoskeleton. 78 (3), 111-125 (2021).
- Wagoner, J. A., Dill, K. A. Evolution of mechanical cooperativity among myosin II motors. Proceedings of the National Academy of Sciences of the United States of America. 118 (20), 2101871118 (2021).
- Walcott, S., Warshaw, D. M., Debold, E. P. Mechanical coupling between myosin molecules causes differences between ensemble and single-molecule measurements. Biophysical Journal. 103 (3), 501-510 (2012).
- Stewart, T. J., Murthy, V., Dugan, S. P., Baker, J. E. Velocity of myosin-based actin sliding depends on attachment and detachment kinetics and reaches a maximum when myosin-binding sites on actin saturate. Journal of Biological Chemistry. 297 (5), 101178 (2021).
- Hilbert, L., Cumarasamy, S., Zitouni, N. B., Mackey, M. C., Lauzon, A. M. The kinetics of mechanically coupled myosins exhibit group size-dependent regimes. Biophysical Journal. 105 (6), 1466-1474 (2013).
- Debold, E. P., Walcott, S., Woodward, M., Turner, M. A. Direct observation of phosphate inhibiting the Force-generating capacity of a miniensemble of myosin molecules. Biophysical Journal. 105 (10), 2374-2384 (2013).
- Kaya, M., Higuchi, H. Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science. 329 (5992), 686-689 (2010).
- Pertici, I., et al. A myosin II nanomachine mimicking the striated muscle. Nature Communications. 9 (1), 1-10 (2018).
- Cheng, Y. S., De Souza Leite, F., Rassier, D. E. The load dependence and the force-velocity relation in intact myosin filaments from skeletal and smooth muscles. American Journal of Physiology - Cell Physiology. 318 (1), 103-110 (2020).
- Stam, S., Alberts, J., Gardel, M. L., Munro, E. Isoforms confer characteristic force generation and mechanosensation by myosin II filaments. Biophysical Journal. 108 (8), 1997-2006 (2015).
- Rastogi, K., Puliyakodan, M. S., Pandey, V., Nath, S., Elangovan, R. Maximum limit to the number of myosin II motors participating in processive sliding of actin. Scientific Reports. 6, 1-11 (2016).
- Debold, E. P., Patlak, J. B., Warshaw, D. M. Slip sliding away: Load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophysical Journal. 89 (5), 34-36 (2005).
- Albert, P. J., Erdmann, T., Schwarz, U. S. Stochastic dynamics and mechanosensitivity of myosin II minifilaments. New Journal of Physics. 16, (2014).
- Erdmann, T., Schwarz, U. S. Stochastic force generation by small ensembles of myosin II motors. Physical Review Letters. 108 (18), 1-5 (2012).
- Guo, B., Guilford, W. H. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motility and the Cytoskeleton. 59 (4), 264-272 (2004).
- Miller-Jaster, K. N., Petrie Aronin, C. E., Guilford, W. H. A quantitative comparison of blocking agents in the in vitro motility assay. Cellular and Molecular Bioengineering. 5 (1), 44-51 (2012).
- Mansoon, A., Balaz, M., Albet-Torres, N., Rosengren, K. J. In vitro assays of molecular motors -- impact of motor-surface interactions. Frontiers in Bioscience. 13, 5732-5754 (2008).
- Persson, M., et al. Heavy meromyosin molecules extending more than 50 nm above adsorbing electronegative surfaces. Langmuir. 26 (12), 9927-9936 (2010).
- Kron, S. J., Spudich, J. A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proceedings of the National Academy of Sciences of the United States of America. 83 (17), 6272-6276 (1986).
- Yanagida, T., Nakase, M., Nishiyama, K., Oosawa, F. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature. 307 (5946), 58-60 (1984).
- Tsuda, Y., Yasutake, H., Ishijima, A., Yanagida, T. Torsional rigidity of single actin filaments and actin-actin bond breaking force under torsion measured directly by in vitro micromanipulation. Proceedings of the National Academy of Sciences of the United States of America. 93 (23), 12937-12942 (1996).
- Stewart, T. J., et al. Actin sliding velocities are influenced by the driving forces of actin-myosin binding. Cellular and Molecular Bioengineering. 6 (1), 26-37 (2013).
- Harada, Y., Sakurada, K., Aoki, T., Thomas, D. D., Yanagida, T. Mechanochemical coupling in actomyosin energy transduction by in vitro movement assay. Journal of Molecular Biology. 216 (1), 49-68 (1990).
- Fordyce, P. M., Valentine, M. T., Block, S. M. Advances in surface-based assays for single molecules. Single-Molecule Techniques: A Laboratory Manual. , 431-460 (2008).
- Ozeki, T., et al. Surface-bound casein modulates the adsorption and activity of kinesin on SiO2 surfaces. Biophysical Journal. 96 (8), 3305-3318 (2009).
- Neuman, K. C., Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nature Methods. 5 (6), 491-505 (2008).
- Neuman, K. C., Block, S. M.
- Thoresen, T., Lenz, M., Gardel, M. L. Thick filament length and isoform composition determine self-organized contractile units in actomyosin bundles. Biophysical Journal. 104 (3), 655-665 (2013).
- Matusovsky, O. S., et al. Millisecond conformational dynamics of skeletal Myosin II power stroke studied by high-speed atomic force microscopy. ACS Nano. 15 (2), 2229-2239 (2021).
- Reinemann, D. N., et al. Collective force regulation in anti-parallel microtubule gliding by dimeric Kif15 kinesin motors. Current Biology. 27 (18), 2810-2820 (2017).
- Cordova, J. C., et al. Bioconjugated core-shell microparticles for high-force optical trapping. Particle and Particle Systems Characterization. 35 (3), 1-8 (2018).
- Reinemann, D. N., Norris, S. R., Ohi, R., Lang, M. J. Processive Kinesin-14 HSET exhibits directional flexibility depending on motor traffic. Current Biology. 28 (14), 2356-2362 (2018).
- Forth, S., Hsia, K. C., Shimamoto, Y., Kapoor, T. M. Asymmetric friction of nonmotor MAPs can lead to their directional motion in active microtubule networks. Cell. 157 (2), 420-432 (2014).
- Shimamoto, Y., Kapoor, T. M. Analyzing the micromechanics of the cell division apparatus. Methods in Cell Biology. 145, 173-190 (2018).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Thoresen, T., Lenz, M., Gardel, M. L.
- Murrell, M., Thoresen, T., Gardel, M.
- Weirich, K. L., Stam, S., Munro, E., Gardel, M. L. Actin bundle architecture and mechanics regulate myosin II force generation. Biophysical Journal. 120 (10), 1957-1970 (2021).
