To illustrate how these protocols, ensure optimal retinal preservation for IHC, we probed retinal sections from P28 WT mice (C57BL/6N) with antibodies to rhodopsin (a photoreceptor marker)8, glutamic acid decarboxylase 65 (Gad65, an amacrine cell marker)9, glutamine synthetase (GS, a Müller cell marker)10, and calbindin (a horizontal cell marker)11 (Figure 4). IHC indicate that our protocols consistently yield high quality and well-preserved retinal sections. Notably, the rhodopsin-rich inner and outer segments remain vertical and intact with little to no separation from overlying the retinal pigmented epithelium (RPE) (Figure 4A). In addition, Müller cells remain well-preserved with soma properly aligned and cytoplasmic processes that span from the ganglion cell layer (GCL) to the outer nuclear layer (ONL) (Figure 4B).
Interneurons, such as Gad65-positive amacrine cells, also remain properly stratified within the INL and inner plexiform layer (IPL) (Figure 4B) while well-defined horizontal cells are detectable at the INL and outer plexiform layer (OPL) border (Figure 4C). Collectively, these data demonstrate that our method preserves retinal cell and tissue integrity, from photoreceptors to ganglion cells
To facilitate transverse sectioning, it is important to meticulously orientate the optical cup in the cryo-mold prior to freezing. Misoriented retinas, poor sectioning technique or sectioning with dull or damaged microtome knives can cause artifacts such as retinal tissue tears, distortions, and cellular displacement, as in Figure 4. Examples of section artifacts are shown in Figure 4D-F. In Figure 4D poor sectioning and alignment lead to photoreceptor inner and outer segments distortion (Figure 4D) and small tissue tears (Figure 4E, arrow). In another example, poor tissue orientation prior to section leads to sub-optimal alignment of Muller cell soma and cytoplasmic processes Figure 4E. Figure 4F shows how poor sectioning likely caused by a dull microtome knife caused horizontal cells to aberrantly smear from the OPL to the GCL. Thus, precise attention must be applied to the execution of each step outlined in the protocol to ensure the highest quality and reproducibility of IHC data.

Figure 1: Tools for mouse eye dissection, embedding, and IHC. (A) Cauterizer; (B) Dissection tools from left to right: Dumont #5/45 curved forceps, Curved scissors, and Dumont #5 thin forceps; (C) 35 mm dissection dish (arrow points to dissecting chamber); (D) Dissecting microscope; (E) Frozen tools from left to right: Insulated cooler, stainless steel beaker for isopentane bath , stainless steel liquid N2 bath; (F) titanium probe; (G) Slide Rack and Coverplate for IHC. Please click here to view a larger version of this figure.
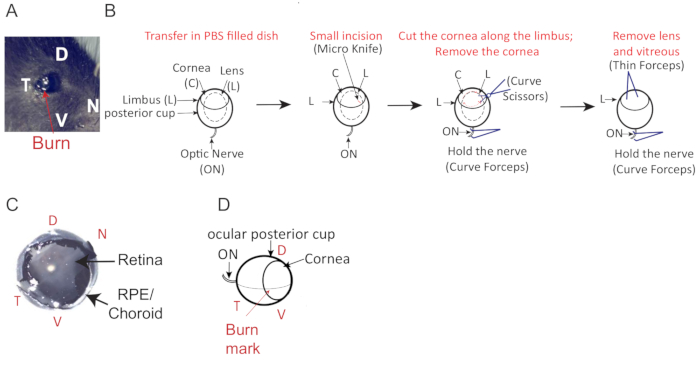
Figure 2: Mouse eye cup orientation and description. (A) Temporal burn mark location (red arrow) to facilitate eye orientation (dorso = D, ventral = V, temporal = T, nasal = N). (B) Schematic for mouse eye dissection. A small incision is made at the burn mark, perpendicular into the cornea and just above the limbus using a fine scalpel (Micro knife). Cut around the cornea to separate the anterior and posterior part of the eye. (C) Eye cup with an intact retina. (D) Dorso-ventral orientation of mouse eye with the optic nerve (ON), lens (L), cornea (C) the limbus (OS). Please click here to view a larger version of this figure.

Figure 3: Mouse eye embedding. (A) Dorso-ventral orientated mice eye cup in cryomold filled with OCT, (B) Frozen mouse retinal cup in annotated cryomold. (C) Cryostat. Please click here to view a larger version of this figure.

Figure 4: Mouse retina immunohistochemistry.(A-C) Good quality and (D-F) poor quality retinal sections from P28 WT were labeled with antibodies to the rhodopsin (A, C) amacrine marker GAD65 (B, E) and the Müller cell marker glutamine synthetase (GS; B, E), and horizontal cell marker calbindin (C, F). White arrows point to tissue tears and aberrant smearing of the tissue. Scale bar, 20 µm. Nuclei labeled with Hoechst 33342 (blue). Please click here to view a larger version of this figure.
