

Summary
Eine robuste Art und Weise die neuronale Lawinen, dh skaleninvarianten räumlich-zeitliche Aktivitätsmuster platzt, was auf kritischen Zustand Dynamik in Kortex zu untersuchen. Lawinen entstehen spontan bei der Entwicklung von oberflächlichen Schichten der kultivierten Cortex, die für die langfristige Messungen der Aktivität ermöglicht mit planaren integrierten Multi-Elektroden-Arrays (MEA) unter genau kontrollierten Bedingungen.
Abstract
Der Kortex ist spontan aktiv, auch in Abwesenheit eines bestimmten Eingang oder Motorleistung. Während der Entwicklung, ist diese Tätigkeit für die Migration und Differenzierung von Cortex Zelltypen und die Bildung neuronaler Verbindungen 1 wichtig. In den reifen Tier, ein Ausdruck der fortlaufenden Tätigkeit der Vergangenheit und den gegenwärtigen Zustand eines Tieres in die Sinnesreize nahtlos integriert sind, um künftige Aktionen zu berechnen. Somit ist ein klares Verständnis von der Organisation der laufenden dh spontane Aktivität eine Voraussetzung für die Cortex-Funktion zu verstehen.
Zahlreiche Aufnahme-Techniken ergab, dass die laufenden Aktivitäten in der Rinde der vielen Neuronen, deren einzelne Aktivitäten vorübergehend zu größeren Veranstaltungen, die im lokalen Bereich Potential (LFP) mit extrazellulären Mikroelektroden detektiert werden können Summe, oder in das Elektroenzephalogramm (EEG), die Magnetoenzephalogramm (MEG ist zusammen ) und das BOLD-Signal von der funktionellen Magnetresonanztomographie (fMRI). Die LFP wird derzeit die Methode der Wahl bei der Untersuchung neuronaler Aktivität Bevölkerung mit hoher zeitlicher und räumlicher Auflösung bei der mesoskopischen Skala (mehrere tausend Neuronen). Bei der extrazellulären Mikroelektroden, lokal Aktivitäten von räumlich benachbarten Neuronen führen schnelle Ausschläge in den LFP bis zu mehreren hundert Mikrovolt synchronisiert. Bei Verwendung einer Anordnung von Mikroelektroden, können die Organisationen wie Ausschläge bequem in Raum und Zeit überwacht werden.
Neuronale Lawinen beschreiben die skaleninvarianten raumzeitliche Organisation der laufenden neuronalen Aktivität im Gehirn 2,3. Sie sind spezifisch für den oberflächlichen Schichten des Kortex als in vitro 4,5 etabliert, in vivo in der narkotisierten Ratte 6 und in der wachen Affen 7. Wichtig ist, deuten sowohl theoretische als auch empirische Studien 2,8-10, dass neuronale Lawinen ein exquisit ausgewogene kritischen Zustand Dynamik des Kortex, die Informationen übertragen und Informationsverarbeitung optimiert anzuzeigen.
Um die Mechanismen der neuronalen Lawine Entwicklung, Wartung und Regulierung Studie, in vitro Vorbereitungen sind sehr nützlich, da sie für stabile Aufnahmen von Lawinen unter genau kontrollierten Bedingungen zu ermöglichen. Das aktuelle Protokoll beschreibt, wie neuronale Lawinen in vitro durch die Nutzung der oberflächlichen Schicht Entwicklung in organotypischen Kulturen Kortex, also Scheibe Kulturen, auf ebenen, integrierter Mikroelektroden-Arrays (MEA, siehe auch 11-14) gewachsen zu studieren.
Protocol
1. Sterile, verschließbares Glasgefäß Kammer mit MEA für Langzeit-Aufzeichnungen
- Threaded Glaszylinder mit Teflon-Kunststoff (Ace Glass), für die sichere und dichte Kultur Kammer Schließung erforderlich ist, werden geschnitten (Aceglass) ca. 2 mm von der Unterseite des den Faden (Abb. 1A, B). Saubere Glas-Ringe durch Spülen mit Wasser (3x) und Kochen für 5 min in 200 Beweis Ethyl-Alkohol, trocknen lassen.
- Aliquot Silizium-Lösung erforderlich, um Glas Ringe MEA Oberfläche befestigen. Mix 15 ml Teile A & B von Sylgard 184 Silikon-Elastomer Kit gründlich, lassen Sie sich für 15 min, um Luftblasen zu entfernen, bewahren in 1 ml Aliquots bei -20 ° C.
- Kleber Glasring der MEA (8x8 Gitter w / interne Masseelektrode, 30 um Elektroden-Durchmesser, 200/100 um inter-Elektrodenabstand für Ratte / Maus) (Abb. 1A, B). Nehmen Sie 1 ml von Silizium (23 ° C) in Spritze mit kleinen Nadel. Silikon auf ungeschliffen Schnittfläche Glasring, Zentrum Glasring auf MEA, gelten eine zusätzliche Schicht von Silizium an der Außenseite des Ringes für eine stärkere Dichtung, lassen Heilung für 1 bis 2 Stunden bei ~ 60 ° C auf einer Heizplatte.
- Sterilisieren MEA Kammer-und Kammermusik Mützen in einer Sterilbank durch 3x in entionisiertem Wasser spülen um 70% Alkohol, gefolgt (3 x; für letzten Spülgang lassen Sie sich für 10 min in Alkohol), gefolgt von 10 min Exposition von Kammer und Deckel innen mit UV-Licht . Autoclave MEA Kammer (120 ° C; nass, 45 min) und trocknen lassen.
- Coat MEA-Oberfläche im Inneren Kultur Kammer mit Poly-D-Lysin. Für neue MEAs, die eher lipophil sind, Mantel durch wiederholtes Tropfen Aspiration der Lösung von der Elektrode Gitter. Bei gebrauchten MEAs, Deckelkammer unten mit einer Lösung, die überschüssige Flüssigkeit absaugen, zu erlauben, unter sterilen Bedingungen in laminaren Haube verdampfen. Bringen Sie Kappe MEA Kammer für die Lagerung und die zukünftige Verwendung zu versiegeln.
2. Zutaten für die Zubereitung und das Wachstum von organotypischen Kulturen Required
- Lösen Sie sterile Agar in 0,9% NaCl, pour in sterile Petrischale (Falcon, 100 x15, ~ 5 mm Höhe), abkühlen lassen und sterilen Verpackung mit Parafilm für die Lagerung. Cut 20 x 10 x 5 mm Blöcke aus massivem Agar für den Einsatz.
- Shop Sekundenkleber, zB Devcon Super Glue II, mit der Verpackung wischte sich mit 70% EtOH vor der Eröffnung, in der Sterilbank, um die Sterilität zu erhalten.
- Bereiten Sie 50% D-Glucose (SIGMA ultra, G7528) durch Zugabe von 40 g Glucose in 40 ml deionisiertem Wasser Kultur (Sigma). Store in 2 ml Aliquots bei -20 ° C.
- 4 ml 50% D-Glucose in 500 ml Balanced Salt Gey-Lösung und Kälte zu Matsch (Mischung aus Flüssigkeit / Eiskristalle) im Gefrierschrank vor dem Gebrauch.
- Rekonstituieren Huhn Plasma in 5 ml deionisiertem Wasser Kultur (shake sanfte, Vermeidung der Bildung von Blasen), lassen Sie für 5 niederlassen - 10 min, leicht schwenken, und Dekantat den klaren Inhalt in eine sterile Petrischale. Sterile-Filter (0,22 um Porenfilter; protein)-Plasma-Lösung, Aliquot 350 ul in Kryoröhrchen (NuncTM), bei -20 ° C.
- Rekonstituieren Thrombin aus bovinem Plasma entsprechend, steril-Filter (0,22 um Porenfilter), 40 ul Aliquot in Kryoröhrchen (NuncTM), bei -20 ° C. Für funktionierende Lösung, verdünnte 40 pl der Thrombin-Lösung in 375 ul einer ausgewogenen Salz Gey-Lösung w / D-Glucose.
- Bereiten Sie 400 ml Kulturmedium durch Mischen von 100 ml Pferdeserum, 200 ml Basal Medium Eagle, 100 ml Hanks Salzlösung auf die 4 ml 50% Glucose und 2 ml 200 mM L-Glutamin hinzugefügt werden. Kann gespeichert werden 4 bis 8 Wochen in 100 ml Flaschen PYREX bei 4 ° C.
- Bereiten Mitose-Hemmer durch Mischen 0,3 mM Uridin, 0,3 mM ARA-C Cytosin-β-D-arabinofuranosid und 0,3 mM 5-Fluor-2'-desoxyuridin, Sterilfilter, Aliquot 200 ul und bei -20 ° C für 6 - 12 Monate.
3. Cortex und ventralen Tegmentum (VTA) Gewebedissektion (Zeit: <1 Stunde)
- Verfahren ergibt Kortex und VTA Gewebeschnitten für ~ 12 Co-Kulturen von Ratten oder Mäusen, und befindet sich in einer Sterilbank unter sterilen Bedingungen hergestellt. Insgesamt benötigte Zeit für die Gewebe-Sammlung soll als 1 Stunde sein.
- Nehmen Sie gesund, gut ernährt Welpen (Wurfgröße ~ 10; Vorhandensein einer abdominalen Milch vor Ort ") bei 1 bis 2 postnatalen Tage (PND). Halten Sie einen Welpen sanft an der Schnauze, damit sie frei hängen und schnell an der Basis des Halses mit einer scharfen Schere zu enthaupten.
- Für Gehirn entfernen, Haut entfernen (zwei seitlichen Scherenschnitt), schneiden Schädel öffnen mit feinem Auge Schere (1 sagittalen Mittellinie geschnitten, 1 koronalen Schnitt bei Cortex / Cerebellum-Kreuzung). Blättern Sie zurück alle 4 Schädel Klappen. Mit einem geschärften Spatel, frontal Schnitt durch den Riechkolben, vorab Spachtel kaudal unterhalb des Gehirns. Heben Sie das Gehirn aus dem Schädel und ließ es in sterile, gekühlt, Gey-Lösung für die schnelle Kühlung und Zwischenlagerung gleiten. Wiederholen Sie die Schritte 3,2 bis 3,3 für 2 weitere Köpfe (Gesamtdauer: <20 min).
- Um VTAGewebe, übertragen Sie die Gehirne auf eine sterile, trockene Petrischale mit einem kleinen Spachtel. Weitere entfernen überschüssige Flüssigkeit durch sanftes Gleiten jedes Gehirn etwa 1 cm seitlich. Entfernen Hirnstamm durch einen koronalen, vertikalen Schnitt auf dem Niveau des Kleinhirns mit einer Rasierklinge.
- Kleber-Agar-Block auf Montage-Disc für die mechanische Stabilisierung des Gehirns während Slicing Verfahren. Legen Sie eine dünne Linie von Sekundenkleber wenige Millimeter vor dem Agar-Block auf der Festplatte (Vermeidung von Leim Berühren der Agar).
- Mit einem kleinen Spatel, Transfer und montieren jedes Gehirn, frontalen Pol nach unten. Stellen Sie sicher, dass frontal Pole Montage Scheibe geklebt und das ventrale Seiten berühren die Agar ohne Klebereste, um die ordnungsgemäße mechanische Stabilisierung während des Schneidens und easy lift-off der geschnittenen Scheiben. Erzielen
- Vorsichtig tauchen und sichere Montage Festplatte mit Gehirn Montage in einem Vibratom Fach (zB Leica VT1000) mit einer sterilen, gekühlt Gey-Lösung gefüllt. Mit einem sorgfältig gereinigt Rasierklinge (90% EtOH), schneiden koronalen Schichten des Mittelhirns bei höchsten Schwingungsfrequenz und relativ niedrige Fahrgeschwindigkeit bei einer Dicke von 400 500 um. Mit einer umgekehrten Pasteur-Pipette mit Saug-Glühbirne, Transfer und sammeln Scheiben mit dem VTA in 35 x 10 mm Petrischalen mit sterilen, gekühlt Gey-Lösung gefüllt (Abb. 1C; siehe auch koronalen Platte 18 bis 20 auf E22 in 15).
- Für Kortex Abschnitte, wiederholen Sie die Schritte 3,2 bis 3,6, sondern gelten vertikalen Schnitt zwischen Kortex und Kleinhirn, und montieren Vorderhirn mit frontalen Pol nach oben. Etwa 3 koronalen Scheiben (350 mu m Dicke) ab auf der Ebene des Striatum sind für zukünftige Cortex Dissektion gesammelt.
- Mit einem Mikro-Messer von gebrochenen Rasierklingen gemacht, sezieren ~ 2 mm breiten Frontalschnitt des frontalen Kortex und des Mittelhirns die Bereiche mit der VTA (Abb. 1C) unter einem Stereomikroskop. Sammeln Gewebeschnitten separat in kleinen Gerichten (z. B. Kammer Dias) mit gekühltem Gey-Lösung gefüllt.
4. Montage Cortex und VTA Gewebeschnitten auf der MEA (Zeit: <1 Stunde)
- Position MEA bei Raumtemperatur unter einem Stereomikroskop mit dem Elektroden-Array im Fokus. Center eine 25 ul Tropfen Plasma auf die saubere, staubfreie und sterile Elektroden-Array-Matrix. Mit kleinen Spachteln, sorgfältig schieben Sie einen Kortex und VTA Abschnitt in das Plasma Tropfen.
- Legen MEA auf Kühlplatte, konzentrieren Sicht ruhen lassen ~ 15 s, dann fügen Sie 25 ul von Thrombin in das Plasma Tropfen. Mit dem Thrombin Pipettenspitze vorsichtig die Plasma / Thrombin-Gemisch mit kleinen kreisenden Bewegungen über die MEA verteilt. Berühren Sie nicht die spröde Elektroden-Array direkt. Vorsichtig Position der Hirnrinde auf das Array mit dorsalen Grenze entlang der zweiten Elektrode Zeile des Arrays. Auf diese Weise wird die Entwicklung von oberflächlichen Schichten schließlich decken die Erinnerung an die Reihe. Die VTA ist benachbart zu dem ventralen Rand des Kortex Abschnitt (Abb. 1D).
- Cap und locker in der Nähe der MEA Kammer mit hoher Luftfeuchtigkeit zu halten, während der MEA / Kultur Montage sitzt ~ 5 min in die Haube bei Raumtemperatur für Plasma / Thrombin Koagulation zu ermöglichen. In der Zwischenzeit, wiederholen Sie Schritt von 4,1 bis 4,3 für 3 weitere Kulturen.
- Vorsichtig 600 ul Kulturmedium in kleine Tröpfchen, die Kultur-Kammer unter Verwendung einer 1 ml-Spritze mit 25 x 5 / 8 Nadel.
- Verschließen Sie die MEA Kammer und Ort MEA / Kultur Montage auf dem schaukelnden Ablage in den Inkubator (Abb. 1B). Zur Beschleunigung des Verfahrens, 3 bis 4 MEAs kann in überlappenden Sequenzen zusammengestellt werden. Montagezeit für 12 MEAs sollte <1 Stunde sein.
- Nach 2 Tagen in vitro (DIV), mit 10 ul der Mitose-Hemmer. Refresh Kulturmedien von 60% bei 4 DIV und alle 4 Tage danach.
5. Elektrophysiologische Aufzeichnung und Stimulus-Generation
- Um die Beziehung zwischen signifikante Ausschläge in den lokalen Bereich Potential (LFP) und die Tendenz der Neuronen Aktionspotenziale Brand zu etablieren, nach ca. 1 Woche 5,6 Rekord spontane Aktivität bei 24 kHz für ~ 10 min von jeder Elektrode des MEA (Hardware : MEA1060 w / Ausblendschaltung, x1200 gewinnen, 12 bit A / D, Bereich 0-4096 mV, mehrkanaligen Systemen, Software: MC_Rack). Ground ist entweder über den internen Masseelektrode vorgesehen, oder extern durch Zugabe einer Ag / AgCl-Halbzelle.
- Trennen Sie die LFP mit einem Bandpass-Filter von 1 bis 200 Hz aus extrazellulären Spike-Aktivität (Bandpass 300 - 3.000 Hz). Spike-Aktivität kann weiter in Einzel-und Multi-Unit-Tätigkeit, bei der off-line spike Sorter (zB Plexon Inc.) klassifiziert werden. Berechnen Spike ausgelöst Durchschnittswerte für jede Elektrode. Für Kortex Kulturen, werden die meisten Durchschnitte negativen LFP Ausschläge (nLFP) als die bevorzugte Zeit der neuronalen Spikes in die Kultur zu identifizieren.
- Berechnen Sie für jede Elektrode eine Schwelle von -3 Standardabweichungen der Lärm (SD) von der LFP Spuren (Abb. 2)Bestimmen Stoßzeiten und Amplituden nLFPs, dass Cross Schwelle (Abb. 2B, C). Wählen Sie eine Zeit bin At (zB zwischen 2 bis 8 ms) und identifizieren raumzeitlichen nLFP Cluster auf dem Array durch die Verkettung nLFPs aus allen Elektrode, die in der gleichen aufeinanderfolgenden Mal Turbinen der Länge At (Abb. 2D; für Details 2,4 s. , 5).
- Um neuronale Lawinen zu identifizieren, berechnen Sie die Größe der einzelnen nLFP Cluster, z. B. Anzahl der aktiven Elektroden oder die Summe der nLFP Amplituden, konstruieren eine Größe Histogramm und Grundstück in doppelt-logarithmischen Koordinaten. Für neuronale Lawinen, folgt die Größenverteilung einem Potenzgesetz durch eine gerade Linie in doppelt-logarithmischen Koordinaten 2 (Abb. 2E, F) angenähert. Siehe 16 für statistische Tests auf Macht Gesetze.
- Elicit evozierte Potentiale im Gewebe durch die Auswahl einer Elektrode, durch die Strom-gesteuerten Reize mit einer Amplitude von S aufgetragen (Stimulus-Generator STG 1008, Multichannel-Systeme) sind. Zur Reduzierung der Elektroden beschädigen, verwenden Sie einen Bereich beschränkt, ladungsneutrale Stimulation der einzelnen Schocks mit bipolaren Rechteck-Wellenform: 50 ms mit einer Amplitude-S, gefolgt von 100 &mgr; mit Amplitude + S / 2 und S zwischen 10 bis 200 uA. Siehe Bedienungsanleitung für weitere Details.
- Zur Erfassung der Dynamik 9, Rekord Stimulus aufgezeichnet LFP Reaktionen bei 4 kHz Abtastrate auf allen Elektroden nach 500 ms nach der Stimulation. Verwenden Sie Blanking-Schaltung (Multi-Kanal-Systeme), die den Kopf der Bühne Verstärker trennt während der Stimulation auf einen Reiz Artefakte zu reduzieren und Vorverstärker Sättigung zu verhindern.
6. Repräsentative Ergebnisse:
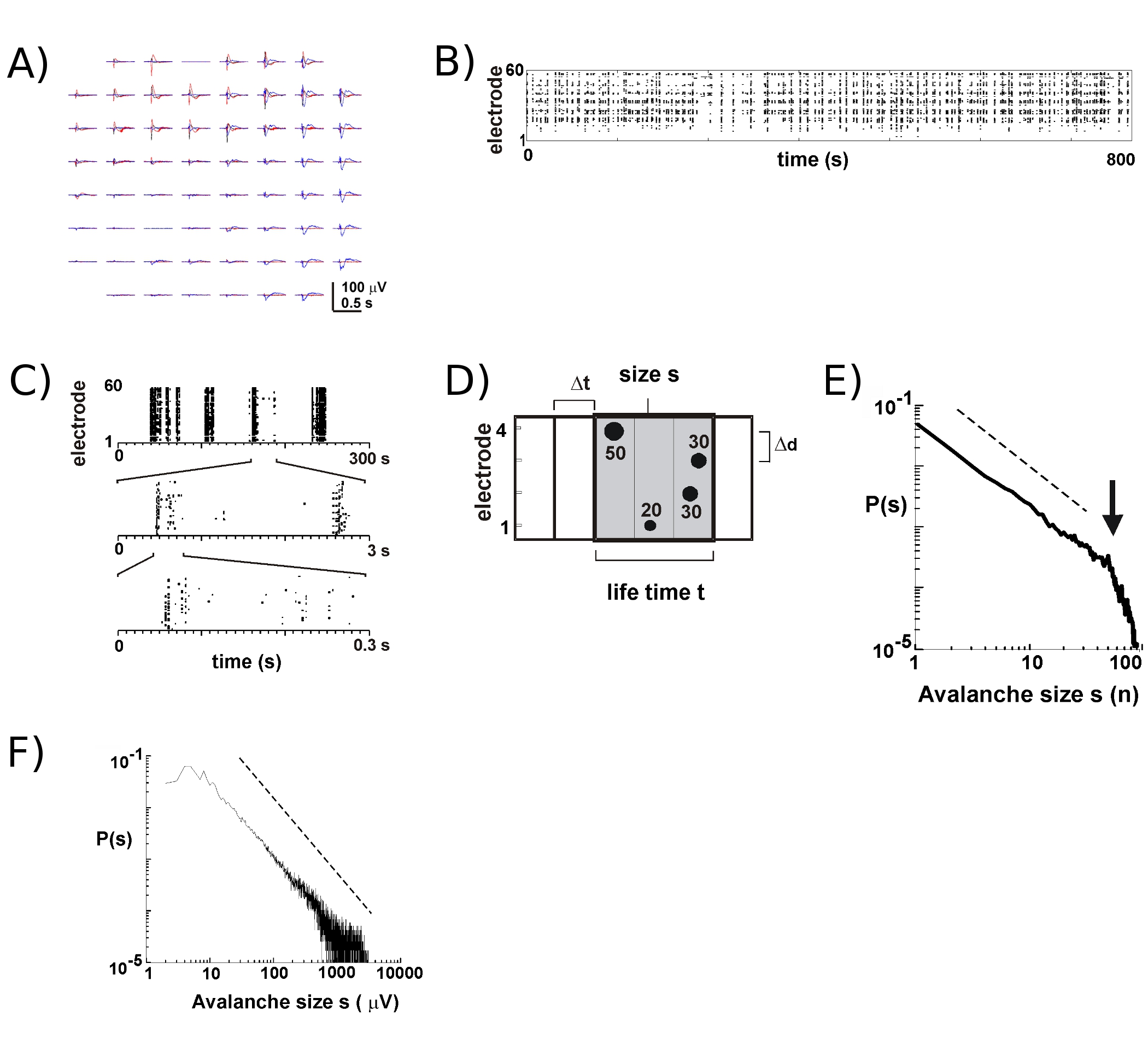
Mit neuen MEAs über 8 bis 9 von 10 Kulturen werden für viele Wochen überleben. Die meisten unserer langfristigen Aufzeichnungen erfolgen im Inkubator in Kulturmedium, die uns auf die Entwicklung der einzelnen Kulturen im Laufe von vielen Wochen 5 verfolgen können. Basierend auf unseren Experimenten können LFP-Aufnahmen zuverlässig mit MEAs für mehr als 100 Kultur Tagen verwendet eingeholt werden. Im Gegensatz dazu ist extrazellulären Spike-Aktivität mehr zuverlässig mit relativ neue MEAs (<40 Kultur Tage) gemessen. In einem typischen Experiment, übertragen wir eine MEA aus der Ablage (Abb. 1B, rechts), um das Fach mit dem Kopf der Bühne befestigt (Abb. 1B, links) halten die Kultur abgedichtet. Für die Hirnrinde 5, Cortex-VTA Co-Kulturen 6, sowie in der narkotisierten Ratte 6 und die wach Affen in vivo 7, neuronale Aktivität während der neuronalen Lawinen in oberflächlichen Schichten tritt vorwiegend in der Nähe der Spitze negativen Auslenkung der LFP (nLFP ). So kann die räumlich-zeitliche Organisation der lokal synchrone neuronale Gruppen, die durch die Messung des Auftretens von nLFPs in Raum und Zeit auf dem Array 17 geschätzt werden.
Aktivität auf dem MEA neigt dazu, in zeitlichen Clustern entstehen, ist, dass Aktivität an einer Elektrode durch Aktivität an anderen Standorten begleitet. Typische Verläufe der LFP während einer solchen Tätigkeit Perioden sind in Abbildung 2a dargestellt, um über Plotten 3 Cluster auftretenden mehrere Sekunden auseinander. Für jedes Cluster können negative Feld Ausschläge an mehreren Elektroden innerhalb eines Fensters von 1 zu sehen s. Beim Extrahieren nLFP Spitzen, die einen Schwellenwert von mehreren negativen SD Kreuz ist die Aktivität in Form von nLFP Spitzenzeiten komfortabel visualisiert in einem Raster, in der "Säulen" der Punkte gibt in der Nähe deckungsgleich nLFPs an verschiedenen Elektroden (Abb. 2B). Die raum-zeitliche Organisation dieser Aktivität ist ziemlich komplex; 'Spalten', die mehr oder weniger homogenen bei niedrigen zeitlichen Auflösung erscheinen, werden von getrennten Clustern auf höhere zeitliche Auflösung und so weiter (Abb. 2C) zusammen. In der Tat ist die Entstehung von räumlich-zeitlichen nLFP Clustern hoch in kortikalen Netzwerken organisiert. Genauer gesagt, ist die Organisation skaleninvarianten für neuronale Lawinen. Dies wird durch die Berechnung der Wahrscheinlichkeit von Cluster-Größen zu einem bestimmten zeitlichen Auflösung At demonstriert. Hier sind Cluster von nLFPs, dass in der gleichen oder aufeinanderfolgenden Zeitintervallen (Abb. 2D) auftreten zusammen. Wenn die Größe eines solchen Clusters in der Gesamtzahl der nLFPs pro Cluster, oder integriert nLFP Amplituden pro Cluster exprimiert wird, zeigt die Cluster-Größe Distributionen einem Potenzgesetz, deren Steigung hat sich gezeigt, auf -1,5 2,4,5,7 werden ( Abb.. 2E, F). Beachten Sie, dass diese Verteilung ein Scale-invariant Bestellung von Cluster-Größen, die das Verhältnis der Größen S bis KXS, wobei k ein konstanter Faktor ist identifiziert, ist k -1,5, die unabhängig von s ist Diese Kraft Gesetzes Organisation ist unabhängig von der Größe des Arrays 2, Ausschläge der zeitlichen Auflösung At 2, und die Schwelle zur erheblichen nLFP identifizieren 7. Da nLFP Amplitude skaliert mit neuronalen Gruppe Größe 7, spiegelt die skaleninvarianten Organisation nLFPs ein Scale-invariant, dh fraktale, Bestellung of lokal neuronalen Gruppen, dass alle Größen sind synchronisiert.

Abbildung 1. (A) Seitenansicht und Draufsicht der MEA mit Gewinde Glasring montiert, und die entsprechenden cap. (B) Innenansicht des Inkubators. Links: headstage Halterung ermöglicht für die Aufnahme von einer einzigen Kultur unter Inkubator Zustand. Rechts: Tray hält zahlreiche MEAs für Kultur Wachstum. Side Räder: Schrittmotor gesteuert rocking Gerät für wechselnde untergetaucht und Atmosphäre ausgesetzt Phase für Kultur das Wachstum notwendig. (C) Schematische Darstellung der koronalen Ratte Scheiben für Cortex-VTA Co-Kulturen verwendet. Cortex Abschnitte (links) und Mittelhirn Abschnitte (Mitte, rechts) mit dem VTA ventralen Tegmentum (VTA; grau) sind durch Schneiden entlang der gestrichelten Linien erhalten. CTX: Cortex, wm: weiße Substanz; cpu: striatum; vta: Pons: pontine Bereich. Siehe auch entsprechende koronalen Platten 8, 18 und 20 um 15. (D) Placement und das Wachstum von einem einzigen Cortex-VTA Co-Kulturen auf der MEA und ihre Entwicklung in den ersten 9 DIV in der Kultur. Beachten Sie die Verflachung der Kultur und deren schrittweise Ausweitung auf dem Array. Reflektierende Gewebeteile zeigen entarteten Zellen und Gewebereste. Gesundes Gewebe ist undurchsichtig und grau unter Durchleuchtung mit sichtbarem Licht.

Abbildung 2. Neuronale Lawinen in kortikalen organotypischen Kulturen. (A) Overplot drei Zeitraum von spontaner Aktivität auf dem Array von mehreren Sekunden getrennt. Beachten Sie, dass jede Aktivität Zeitraum von negativen LFP Ausschläge auf vielen Elektroden auf dem Array (jede Farbe Etiketten eine Aktivität Periode) besteht. (B) Negative Spitzenzeiten nLFPs von jeder Elektrode werden in einem Raster von Aktivitäten zusammengestellt. "Column' Strukturen angegeben Zeiten in der Nähe synchrone Aktivität. (C) Beachten Sie, dass Spalten, die sehr an einer Zeitskala angezeigt werden synchronisiert von mehreren Spalten in den höheren zeitlichen Skalen (3 zeitlichen Skalen dargestellt) bestehen. (D) Schematische Darstellung der neuronalen Lawine Algorithmus. Auf einer 2 x 2 Elektroden-Array Peak-Zeit und Amplitude der negativen Ausschläge LFP (nLFP) Überqueren einer Schwelle von-x SD des Lärms identifiziert. Räumlich-zeitliche Organisation der nLFPs ist in der Reihe der aktiven Zeit bins der Breite At geclustert. Die Größe eines Clusters wird entweder durch die Anzahl der aktiven Zentren, dh Elektroden mit nLFP identifiziert (s = 4) oder die integrierte Summe nLFP Amplituden (s = 130 uV). Die Lebensdauer wird in Vielfachen von At gemessen. (E, F) Power Gesetz in Cluster-Größenverteilung identifiziert Cluster als neuronale Lawinen. Beachten Sie, dass die Wahl eines bestimmten interelectrode Entfernungen für das Array (hier 200 pm) Dd eine besondere At, bei dem die Dynamik beobachtet werden sollte einführt. Genauer gesagt, in welchem Verhältnis Dd / At nähert sich der durchschnittliche Ausbreitungsgeschwindigkeit im Netz, bei der die Steigung α der Macht Recht ist annähernd -1,5 für die neuronale Lawinen 2,4,5. Bitte klicken Sie hier, um eine größere Version der Abbildung 2 zu sehen.
Discussion
1. Technische Fragen:
- Sterile Technik. Die Aufstellung von MEAs und Kultur Vorbereitung sind alle in einer Sterilbank unter sterilen Bedingungen durchgeführt. Antibiotika, die neuronale Aktivität beeinflussen, sind nicht zu jeder Zeit während der Vorbereitung und Kultur eingesetzt.
- Plasma / Thrombin Koagulation und Gewebe Einhaltung der MEA. Tissue Überleben auf dem MEA erfordert eine sorgfältige Balance zwischen der Zeit, für Plasma / Thrombin Koagulation und die Zeit des Gewebes an der Luft erforderlich. Eine kurze Gerinnungszeit Risiken vorzeitigen Ablösung von Abschnitten aus der MEA, während längerer Atmosphäre löst Gewebedegeneration. Da die Stärke des Thrombin-Lösung bestimmt die Geschwindigkeit der Koagulation ist es ein sehr wichtiger Parameter für die erfolgreiche Anbringung, gesunde Kulturen der MEA-Oberfläche. Wir erzielen die besten Ergebnisse mit 1000 Einheiten (1KU; 1 NIH-Einheit = 0,324 ± 0,073 mg). Wichtig ist, dass unvollständige Vermischung der Plasma / Thrombin-Lösung resultiert in räumlich heterogenen Koagulation Förderung Plasma-Brüche während der Kultivierung. Diese Plasma-"Löcher" erheblich beeinträchtigen Kultur Gesundheit und lösen Sie die Kultur von Mikroelektroden, damit Abstriche bei der Qualität der elektrophysiologischen Aufzeichnung. Arbeiten mit Kühlplatten während der MEA / Gewebe Montage verlangsamt die Blutgerinnung und ermöglicht eine homogene Durchmischung des Plasmas / Thrombin-Lösung und die richtige Positionierung der Gewebeschnitten.
Auch durch Zugabe von Medium in einzelne Tröpfchen, die Kultur Kammer zu versenken die Kultur nach nur 5 min von Koagulation, reduziert das Risiko von Gewebe Abteilungen aufgrund der Oberflächenspannung. Eine erfolgreiche Kultur wird auf der MEA flach und leicht zu erweitern, welche ein gesundes Wachstum für Wochen, ohne größere Anzeichen von unvollständiger Gewebe-MEA Kontakt oder Gewebedegeneration (z. B. Abb. 1D). - Gewebedissektion. Micro Messer haben unsere Dissektion Prozess verbessert. Wir verwenden doppelte Rasierklingen (Fine Science Tools - zerbrechlich Skalpellklingen - 100050-00), aus denen wir uns trennen ~ 2 mm breite Klingen "mit einer Zange, und halten Sie sie mit einem Skalpell Inhaber. Gewebeproben aus den koronalen Schnitt mit einem glatten, senkrechten festen Bewegung des Messers seziert dabei stark reduziert mechanische Belastung durch Ziehen des Gewebes insgesamt verbessern Kultur Gesundheit.
- Tissue Kühlung. Ordnungsgemäße Kühlung der Scheiben und Gewebeschnitte während der Vorbereitung ist unerlässlich für die Kultur Erfolg. Wir verwenden maßgeschneiderte kalten Platten von Peltier-Elementen unter einer Metallscheibe befestigt gebaut. Die Wärme, die durch das Peltier-Element erzeugt wird, durch kaltes Wasser Perfusion entfernt. Dies reduziert die Vorbereitungszeit und standardisiert Kühlung während jeder Phase der Vorbereitung (Dold Labs & Engineering 131 Plantation Dr. Seguin, TX 78155;. (830) 560-1471)).
- Incubator Zustand. Eine maßgeschneiderte Inkubator mit präzisen internen rocking Bedingungen war entscheidend für unseren Erfolg in der Pflege Scheiben auf MEAs. Basierend auf einem Original in-house design by Multichannelsystems (derzeit nicht im Handel erhältlich), besteht die interne rocking Gerät von Schalen, die mit zwei großen seitlichen Rädern angebracht sind. Schrittmotoren und EDV-Steuerung ermöglicht eine präzise Flugbahn rocking (rocking Winkel, rocking Geschwindigkeit und intermittierenden Pausen). Letztlich müssen Slice Kulturen in die Atmosphäre und Kulturmedium in langsamen Wechsel ausgesetzt werden. Der traditionelle Ansatz besteht darin, Slice Kulturen in engen Röhren, die sich langsam entlang gibt es am längsten Achse zu drehen statt. Dabei spielt langsame Rotation nicht produzieren mechanische Belastung durch die Rotation selbst, und die Rotationsgeschwindigkeit ist hoch genug, um eine optimale Fütterung / Atmung "-Zyklus von etwa 5 erhalten - 10 min Dauer. Je kompakter Innere des MEA Kammer, ihre Gesamtvolumen von ca. 2 ccm, und die kleine Kulturmedium Volumen für mittlere Konditionierung durch das Gewebe selbst erforderlich sind, stellt eine erhebliche Herausforderung. Durch Schütteln der MEA zwischen ± 70 ° Winkel (Zykluszeit: ~ 200 s), Verlangsamung der rocking Geschwindigkeit wie die Kultur Übergänge zwischen der flüssigen Phase und die Atmosphäre, und die Eindämmung des rocking an den äußersten Winkeln für längere Einwirkung der Atmosphäre hat wesentlich gewesen für die Kultur überleben.
2. Developmental Alter von Cortex Kulturen zu neuronalen Lawinen-Studie
Akute Scheiben von Rattencortex werden in der Regel bei PND 0 Gewinner - 1 und kultiviert für viele Wochen auf der MEA. Frühe Studien haben deutlich gezeigt, dass einzelne Kortex Scheibe Kulturen, nach mehreren Wochen in vitro, pflegen eine geschichtete Struktur mit identifizierbaren Zelltypen, die sich leicht in vivo Zell-Klassen 18,18-21 verglichen werden kann. Das geschichtete Organisation in diesem in vitro-System wurde verwendet, um bequem Thalamus StudieInnervation der Hirnrinde während der Entwicklung 22-24, sowie für den Antrieb subkortikale Strukturen wie das Striatum 25,26. In der Tat ermöglicht die Spezifität bei der Bildung von neuronalen Verbindungen innerhalb und zwischen den Hirnregionen für den Bau des Komplexes in vitro-Systeme, zahlreiche detaillierte Projektionssysteme zurückzuerobern, zB, dass der Kortex-Basalganglien-Schaltung 27-30.
Nach 4 bis 6 Wochen in vitro, single Kortex Scheiben 31 und Cortex Scheiben mit striatum 26 oder Thalamus 32 zeigen spontane co-kultivierten Up-und Down Staaten typischerweise in vivo in der Urethan-narkotisierten Ratten 33 gefunden. Die feine zeitliche Organisation dieser up-Staaten trägt die Handschrift von verschachtelten θ-und γ-Oszillationen weisen auf eine elektrophysiologisch reifen Netzwerk von pyramidalen Neuronen und schnell spiking GABAergen Interneuronen 31. Wichtig ist, dass in der Abwesenheit von Dopamin-D2-Rezeptor-Stimulation, ist die Reifung von Parvalbumin-positive kortikale Interneuronen von ca. 2 Wochen in Cortex Schnittkulturen 34 verzögert. Im Einklang mit diesen Ergebnissen die Entwicklung zeitlichen Verlauf der verschachtelten θ-, β-und γ-Oszillationen ist, dass in vivo trifft zu, wenn Kortex Scheiben sind mit dem ventralen Tegmentum (VTA), die dopaminergen Neuronen projizieren, die enthält co-kultivierten Cortex 6.
Diese Studien zeigen, dass bei der Untersuchung neuronaler Lawinen, die entscheidend davon abhängen, reifen schnell GABAergen Hemmung und sind in oberflächlichen Schichten des Kortex 2,4 gelegen, großer Sorgfalt zu treffen, um eine ordnungsgemäße Reifung der kortikalen Gewebes zu gewährleisten werden muss. Während neuronale Lawinen in einzelnen Kortex Kulturen entstehen über den zeitlichen Verlauf der 2 bis 5 Wochen 4, wenn die eine Entwicklungszeit natürlich, dass die in vivo Entwicklung abgestimmt ist, müssen Kortex Scheiben eine entsprechende Dopamin-Rezeptor-Stimulation, zB durch Co-Kultivierung Kortex Scheiben mit dem VTA 6.
Disclosures
Keine Interessenskonflikte erklärt.
Acknowledgments
Diese Studie wurde von der Abteilung für Interne Research Program (DIRP) des National Institute of Mental Health, National Institutes of Health gefördert.
Materials
| Name | Company | Catalog Number | Comments |
| Integrated planar multielectrode array |  Multi Channel System MCS GmbH Multi Channel System MCS GmbH |
200/30iR-ITO-w/o | Titanium Nitrate (TiN) electrodes (30 mm diameter) have large surface resulting in low impedance ( ~1.5 kΩ at 1 kHz) and excellent wide-band recordings ( w/o -– without ring) |
| Chamber glass | www.aceglass.com | 7620-32 | Threaded glass cylinder |
| Chamber cap | www.aceglass.com | 7622-114 | Plastic cap with Teflon insert |
| Sylgard 184 | World Precision Instruments, Inc. | SYLG184 | Two-part silicone elastomer |
| Poly-D-lysine | Sigma-Aldrich | P6407-5mg | γ-irradiated, lyophilized powder, cell cultured tested. Reconstituted with 5 ml deionised water before use. |
| Gey’s Balanced Salt solution | Sigma-Aldrich | G9779-500mL | sterile filtered and cultured tested |
| chicken plasma | Sigma-Aldrich | P3266-5mL | Lyophilized, reconstitute with 5 ml deionized water before use. |
| thrombin | Sigma-Aldrich | T6634-1KU | from bovine plasma, lyophilized powder form. |
| horse serum | Sigma-Aldrich | H1138-100mL | donor herd, heat inactivated, cell culture tested |
| Basal Medium Eagle | Invitrogen | 21010-046 | 1x, 500 ml - (+) Earle’s Salts, (-) L-glutamine), |
| Hank’s Buffered Saline Solution | Invitrogen | 24020-117 | 500 ml - (+) Magnesium, (+) calcium, w/phenol red) |
| Chamber slides | Lab-Tek | 177429 | |
| Uridine | Sigma-Aldrich | U3003 | |
| ARA-C cytosine-β-D-arabinofuranoside | Sigma-Aldrich | C6645 | |
| 5-fluoro-2’-deoxyuridine | Sigma-Aldrich | F0503 |
References
- Spitzer, N. C. Electrical activity in early neuronal development. Nature. 444, 707-712 (2006).
- Beggs, J. M., Plenz, D. Neuronal avalanches in neocortical circuits. J. Neurosci. 23, 11167-11177 (2003).
- Beggs, J. M., Plenz, D. Neuronal avalanches are diverse and precise activity patterns that are stable for many hours in cortical slice cultures. J Neurosci. 24, 5216-5229 (2004).
- Stewart, C. V., Plenz, D. Inverted-U profile of dopamine-NMDA-mediated spontaneous avalanche recurrence in superficial layers of rat prefrontal cortex. J. Neurosci. 26, 8148-8159 (2006).
- Stewart, C. V., Plenz, D. Homeostasis of neuronal avalanches during postnatal cortex development in vitro. J. Neurosci. Meth. 169, 405-416 (2007).
- Gireesh, E. D., Plenz, D. Neuronal avalanches organize as nested theta- and beta/gamma-oscillations during development of cortical layer 2/3. Proc. Natl. Acad. Sci. U. S. A. 105, 7576-7581 (2008).
- Petermann, T. Spontaneous cortical activity in awake monkeys composed of neuronal avalanches. Proc. Natl. Acad. Sci. U. S. A. 106, 15921-15926 (2009).
- Kinouchi, O., Copelli, M. Optimal dynamical range of excitable networks at criticality. Nature Physics. 2, 348-351 (2006).
- Shew, W. L., Yang, H., Petermann, T., Roy, R., Plenz, D. Neuronal avalanches imply maximum dynamic range in cortical networks at criticality. J. Neurosci. 29, 15595-15600 (2009).
- Shew, W. L., Yang, H., Yu, S., Roy, R., Plenz, D. Information capacity is maximized in balanced cortical networks with neuronal avalanches. J Neurosci. 5, 55-63 (2011).
- Karpiak, V., Plenz, D. Preparation and maintenance of organotypic cultures for multi-electrode array recordings. Current Protocols in Neuroscience. 1, 6-6 (2002).
- Hammerle, H., Egert, U., Mohr, A., Nisch, W. Extracellular recording in neuronal networks with substrate integrated microelectrode arrays. Biosens. Bioelectron. 9, 691-696 (1994).
- Nisch, W., Bock, J., Egert, U., Hammerle, H., Mohr, A. A thin film microelectrode array for monitoring extracellular neuronal activity in vitro. Biosens. Bioelectron. 9, 737-741 (1994).
- Egert, U. A novel organotypic long-term culture of the rat hippocampus on substrate-integrated multielectrode arrays. Brain Res. Protoc. 2, 229-242 (1998).
- Altman, J., Bayer, S. A. Atlas of Prenatal Rat Brain Development. , CRC Press. Boca Raton. (1995).
- Clauset, A., Shalizi, C. R., Newman, M. E. J. Power-law distributions in empirical data. arXiv. , (2009).
- Plenz, D., Thiagarajan, T. C. The organizing principles of neuronal avalanches: cell assemblies in the cortex? Trends Neurosci. 30, 101-110 (2007).
- Cäser, M., Bonhoeffer, T., Bolz, J. Cellular organization and development of slice cultures from rat visual cortex. Exp. Brain Res. 477, 234-244 (1989).
- Cäser, M., Schüz, A. Maturation of neurons in neocortical slice cultures. A light and electron microscopic study on in situ and in vitro material. J. Hirnforsch. 33, 429-443 (1992).
- Götz, M., Bolz, J. Development of vasoactive intestinal polypeptide (VIP)-containing neurons in organotypic slice cultures from rat visual cortex. Neurosci. Lett. 107, 6-11 (1989).
- Götz, M., Bolz, J. Formation and Preservation of cortical layers in slice cultures. J. Neurobiol. 23, 783-802 (1992).
- Bolz, J., Novak, N., Götz, M., Bonhoeffer, T. Formation of target-specific neuronal projections in organotypic slice cultures from rat visual cortex. Nature. 346, 359-362 (1990).
- Bolz, J., Novak, N., Staiger, V. Formation of specific afferent connections in organotypic slice cultures from rat visual cortex cocultured with lateral geniculate nucleus. J. Neurosci. 12, 3054-3070 (1992).
- Novak, N., Bolz, J. Formation of specific efferent connections in organotypic slice cultures from rat visual cortex cocultured with lateral geniculate nucleus and superior colliculus. Eur. J. Neurosci. 5, 15-24 (1993).
- Plenz, D., Aertsen, A. Neural dynamics in cortex-striatum co-cultures. I. Anatomy and electrophysiology of neuronal cell types. Neurosci. 70, 861-891 (1996).
- Plenz, D., Aertsen, A. Neuronal dynamics in cortex-striatum co-cultures. II. Spatio-temporal characteristics of neuronal activity. Neurosci. 70, 893-924 (1996).
- Plenz, D., Kitai, S. T. Organotypic cortex-striatum-mesencephalon cultures: the nigro-striatal pathway. Neurosci. Lett. 209, 177-180 (1996).
- Plenz, D., Kitai, S. T. Up' and 'down' states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex-striatum-substantia nigra organotypic cultures. J. Neurosci. 18, 266-283 (1998).
- Plenz, D., Herrera-Marschitz, M., Kitai, S. T. Morphological organization of the globus pallidus-subthalamic nucleus system studied in organotypic cultures. J. Comp. Neurol. 397, 437-457 (1998).
- Plenz, D., Kitai, S. T. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus [see comments]. Nature. 400, 677-682 (1999).
- Plenz, D., Kitai, S. T. Generation of high-frequency oscillations in local circuits of rat somatosensory cortex cultures. J Neurophysiol. 76, 4180-4184 (1996).
- Klostermann, O., Wahle, P. Patterns of spontaneous activity and morphology of interneuron types in organotypic cortex and thalamus-cortex cultures. Neurosci. 92, 1243-1259 (1999).
- Cowan, R. L., Wilson, C. J. Spontaneous firing patterns and axonal projections of single cortico-striatal neurons in the rat medial agranular cortex. J. Neurophysiol. 71, 17-32 (1994).
- Porter, L. L., Rizzo, E., Hornung, J. P. Dopamine affects parvalbumin expression during cortical development in vitro. J Neurosci. 19, 8990-9003 (1999).

{kind=link}