

Summary
Given that GPCRs are attractive druggable targets, GPCR ligand screening is thus indispensable for the identification of lead compounds and for deorphanization studies. Towards these efforts, we describe PRESTO-Tango, an open-source resource platform used for simultaneous profiling of transient β-arrestin2 recruitment at approximately 300 GPCRs using a TEV-based reporter assay.
Abstract
As the largest and most versatile gene superfamily and mediators of a gamut of cellular signaling pathways, G-protein-coupled receptors (GPCRs) represent one of the most promising targets for the pharmaceutical industry. Ergo, the design, implementation, and optimization of GPCR ligand screening assays is crucial, as they represent remote-control tools for drug discovery and for manipulating GPCR pharmacology and outcomes. In the past, G-protein dependent assays typified this area of research, detecting ligand-induced events and quantifying the generation of secondary messengers. However, since the advent of functional selectivity, as well as an increased awareness of several other G protein-independent pathways and the limitations associated with G-protein dependent assays, there is a greater push towards the creation of alternative GPCR ligand screening assays. Towards this endeavor, we describe the application of one such resource, the PRESTO-Tango platform, a luciferase reporter-based system that enables the parallel and simultaneous interrogation of the human GPCR-ome, a feat which was previously considered technically and economically unfeasible. Based on a G-protein independent β-arrestin2 recruitment assay, the universality of β-arrestin2-mediated trafficking and signaling at GPCRs makes PRESTO-TANGO an apt tool for studying approximately 300 non-olfactory human GPCRs, including approximately 100 orphan receptors. PRESTO-Tango's sensitivity and robustness make it suitable for primary high-throughput screens using compound libraries, employed to uncover new GPCR targets for known drugs or to discover new ligands for orphan receptors.
Introduction
G-protein-coupled receptors (GPCRs) constitute the largest and most diverse family of transmembrane proteins, operating as communication interfaces between a cell and its environment1. The versatility of GPCRs is highlighted by their ability to detect a diverse array of ligands–from neurotransmitters to nucleotides, peptides to photons, and many more–as well as their ability to regulate numerous downstream signaling cascades involved in cellular growth, migration, differentiation, apoptosis, cell firing, etc.2,3. Considering their ubiquity and involvement in a multitude of physiological processes, this receptor family is of utmost therapeutic importance, showcased by the fact that more than a third of currently available prescribed medications target GPCRs4. However, these existing therapeutics only target a small subset of the superfamily (an estimated 10%), and the pharmacology of many GPCRs remains unelucidated. Moreover, more than 100 GPCRs exist as orphan receptors, as they have not been matched with an endogenous ligand5. Thus, GPCR ligand screening is critical in deorphanization and drug development, as it paves the path towards lead discovery and optimization, and possibly to the clinical trial phase.
Methods for GPCR ligand screening have traditionally fallen in one of two categories, G-protein dependent or G-protein independent functional assays6. GPCR signaling is regulated by heterotrimeric G-proteins (Gαβγ), which are activated by the exchange of GTP for GDP bound on the Gα subunit7. Signals from the activated receptor are transduced by G-proteins via secondary messengers, such as cAMP, Calcium, DAG, and IP3, to mediate downstream signaling at downstream effectors8. The nature of the functional consequences of G-protein signaling has been exploited to create cell-based assays that reflect receptor activation. These methods, which measure proximal (direct) or distal (indirect) events in G-protein signaling, are most frequently used for GPCR ligand screening and have been principally employed in deorphanization studies6. Examples of assays that directly measure GPCR-mediated G-protein activation include the [35S]GTPγS binding assay, which measures binding of a radiolabeled and non-hydrolyzable GTP analog to the Gα subunit, and Förster/bioluminescence resonance energy transfer (FRET/BRET, respectively) probes to monitor GPCR-Gα and Gα/Gγ interactions, which have been gaining more traction over the years9,10. Assays that monitor distal events are the most commonly used tools for GPCR profiling; for example, cAMP and IP1/3 assays measure intracellular accumulation of G-protein dependent secondary messengers, whereas [Ca2+] flux and reporter assays involving specific response elements implicated in G-protein activation (CRE, NFAT-RE, SRE, SRF-RE) examine events further downstream the signaling cascade11. While most of the aforementioned assays can be performed at a high-throughput level, are fairly sensitive, and boast certain assay-specific advantages (e.g., discrimination between full/partial agonists, neutral antagonists and inverse agonists in the case of GTPγS binding, or assay functionality on live cells such as [Ca2+] and IP1/3)6, there are unfortunately no existing G-protein dependent methods befitting the interrogation of the entire druggable GPCR-ome. This is largely due to the native coupling of multiple G-protein subfamilies to GPCRs, resulting in signaling at several cascades and the unknown G-protein coupling at orphan GPCRs. To mitigate this issue, assays have been developed to force promiscuous G-protein coupling through a single common signaling read-out, such as cAMP, and Ca2+, albeit most of them are low-throughput12.
An important aspect of the GPCR lifecycle is the termination of G-protein-dependent signaling, which occurs in large part through the recruitment of β-arrestins which induces dissociation of the G-protein, and ultimately desensitizing the receptor, which is targeted for clathrin-coated internalization13. The most ubiquitously expressed isoforms of β-arrestin are the non-visual β-arrestin1 and β-arrestin2, also denoted as arrestin-2 and arrestin-3, respectively14. Enter G-protein independent cell-based assays, which add a new dimension to GPCR ligand screening; receptor trafficking, label-free whole cell, and β-arrestin recruitment assays are all notable examples. GPCR trafficking assays employ fluorophore-labeled ligands or co-internalized antibodies targeting the receptor15, whereas label-free whole cell assays use biosensors which translate cellular changes induced by ligand binding into quantifiable outputs, such as electrical or optical signals16. Notably, quintessential GPCR- β-arrestin interactions fashion the β-arrestin recruitment assay as an attractive tool in the repertoire of functional assays17. The Tango system, first developed by Barnea et al. only a decade ago, involves the introduction of three exogenous genetic elements: a protein fusion consisting of β-arrestin2 with a tobacco etch virus protease (TEVp), a tetracycline transactivator (tTA) that is tethered to a GPCR via a tobacco etch virus protease cleavage site (TEVcs) and is preceded by a sequence from the C-terminus of the V2 vasopressin receptor (V2 tail) to promote arrestin recruitment, and a reporter luciferase gene whose transcription is triggered by the tTA transcription factor translocation to the nucleus, which is freed from the membrane anchoring following β-arrestin2 recruitment (Figure 1)18. Quantitative readings of GPCR activation and β-arrestin2 recruitment can be subsequently determined by reading for luminescence. A notable distinction is that while receptor trafficking and label-free whole cell methods are relatively low-throughput, the Tango has several advantages, including selective read-out that is specific to the target receptor and sensitivity due to signal integration, which make it a suitable candidate for ligand screening on a larger scale18.
In view of these strategic features, Kroeze et al. developed PRESTO-Tango (Parallel Receptor-ome Expression and Screening via Transcriptional Output-Tango), a high-throughput open-source platform that uses the Tango approach to profile the druggable GPCR-ome in a parallel and simultaneous manner19. Exploiting the "promiscuous" recruitment of β-arrestin2 to nearly all GPCRs, PRESTO-Tango is the first-of-its-kind in terms of cell-based functional assays, enabling rapid "first-round" screening of small molecule compounds at almost all non-olfactory GPCRs, including orphans, independent of the G-protein subfamily coupling.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Primary screening: cell culture and plate seeding
- To prepare poly-L-lysine (PLL)-coated plates, dispense 20 μL/well of a 25 μg/mL stock solution of PLL in white or black 384-well optical bottom plates using an electronic multichannel pipette or a reagent dispenser. Incubate the plates at room temperature for 0.5–2 h.
NOTE: If using the black 384-well plates, expect the background signal to be lower compared to the white plates. Black plates are recommended to reduce bleed-through of luminescence between adjacent wells. - To preserve the coated plates and wash off the excess PLL, remove the PLL by flicking it over the sink, tap dry over a paper towel, and add 40 μL/well of diluted 1x solution of antibiotic-antimycotic using an electronic multichannel pipette or a reagent dispenser. Store PLL-coated plates at 4 °C until ready for plate seeding.
- Maintain HTLA cells (kindly provided by Dr. Richard Axel)–a human embryonic kidney cell line (HEK293T) stably expressing β-arrestin2-TEV and tTA-driven luciferase–in complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% of Fetal Bovine Serum, 5% of Bovine Calf Serum, 2.5 μg/mL of puromycin, 50 μg/mL of hygromycin, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified incubator containing 5% CO2.
- Culture HTLA cells in 150 mm dishes and pass cells twice a week at a dilution factor of 1:10, with optimal cell passage number of 5–25. Ensure that a sufficient number of 150 mm dishes are confluent the day of 384-well plate seeding, depending on the scale of the primary screen.
NOTE: Usage of HTLA cells greater than passage 25 may result in reduced viability, yielding suboptimal results. - To seed HTLA cells for the primary screen, gently rinse the confluent 150 mm dish(es) with 1x phosphate-buffered saline (PBS), pH 7.4. Detach cells with approximately 6 mL of 0.05% Trypsin/0.53 mM EDTA, and transfer to a centrifuge tube containing at least equal amount of complete Dulbecco's modified Eagle medium (DMEM) to neutralize the trypsin.
- Spin down HTLA cells at 500 x g for 3 min and resuspend the cell pellet at a density of 0.22 x 106 cells/mL in complete DMEM, omitting the addition of 2.5 μg/mL of puromycin and 50 μg/mL of hygromycin as they can decrease transfection efficacy.
- Incubate the necessary 384-well PLL-coated plates at 37 °C to warm them before seeding cells. Remove the storage solution of 1x antibiotic-antimycotic from the 384-well PLL-coated plate(s) by flicking the plate over the sink and taping it over a paper towel to dry.
- Seed cells into the 384-well PLL-coated plates at a final density of 10,000 cells/well by dispensing 45 μL of the 0.22 x 106 cells/mL HTLA suspension using an electronic multichannel pipet. Incubate plates at 37 °C overnight. If a same-day transfection is preferred, seed cells at a density of 16,000 cells/well and perform the transfection 4 h later.
NOTE: For high transfection efficiency, 50–70% cell confluency is optimal.
2. Primary screening: DNA plate preparation and transfections
- To prepare the 384-well DNA source plate for transfection as shown in Figure 2, distribute the plasmid cDNAs encoding the GPCR-Tango constructs of interest in a 96-well plate, with a different GPCR/well. The plasmid DNA should be suspended in 0.1x Tris-EDTA (TE) buffer at a concentration of 50 ng/μL.
NOTE: The 96-well DNA plates can be sealed and stored at -20 °C, and re-used for multiple screening experiments. All cDNA encoding GPCR-Tango constructs are available commercially (see the Table of Materials) and are cloned in the pcDNA3.1 neomycin plasmid. The PRESTO-Tango GPCR Kit consists of four 96-well plates, which include 80 GPCRs each, a couple of wells with an empty vector as negative controls, and positive control wells that hold the dopamine receptor D2 (DRD2), and wells that carry a plasmid encoding a fluorescent protein (YFP) to track transfection efficiency. - Using a multichannel pipette, manually transfer DNA solution from the 96-well to the a 384-well DNA source plate, adding 10 μL per 384-well. To ensure that each condition of the experiment is assayed in quadruplicate, half of the 96-well DNA plate (rows A-D or E-H) will cover a full 384-well plate by distributing each GPCR in two quadrants (first quadrant = - compound, second quadrant = + compound), such that the same GPCR will be transfected in 8 wells of the 384-well plate (see Figure 2 as a guide).
- Assemble the following transfection reagents needed for calcium phosphate precipitation method, as described by Jordan et al.20: 0.1x TE buffer (1 mM Tris-HCl and 0.1 mM EDTA); 2.5 M CaCl2 solution; 2x Hepes buffer, pH 7.05 (50 mM HEPES, 280 mM NaCl, 1.5 mM Na2HPO4). Sterilize all the solutions by filtration and store at 4 °C. The day of transfection, allow the reagents to reach room temperature before use.
- Dilute the 2.5 M CaCl2 stock solution in 0.1x TE (1:8 dilution) to a final concentration of 0.313 M CaCl2 and vortex. Transfer 40 μL of 0.313 M CaCl2 to the 384-well DNA source plate and mix by pipetting up and down with a hand-held multichannel pipette or an automated benchtop 384-channel pipettor.
- Add 50 μL of 2x Hepes buffer to the 384-well DNA source plate, mix again by pipetting up and down and let stand for 1 min; each 384-well will have an adequate amount of DNA/transfection mixture for the transfection of nine 384-well plates, depending on the number of compounds that need to be tested. Transfer 10 μL of the DNA/transfection mixture from the 384-well DNA source plate to the seeded HTLA cells and incubate the plates overnight at 37 °C.
3. Primary screening: Cell stimulation
- Twenty-four hours later, decant the transfected cell media by gently flicking the 384-well plate over the sink and taping it over a paper towel, or with an aspirator head. Slowly add 40 μL of starving media (DMEM supplemented with 1% dialyzed fetal bovine serum (dFBS) and 1x antibiotic/antimycotic), being careful to avoid touching the cells directly.
- Pipet 20 μL of the compound of interest at a 3x concentration (final concentration of the drug in the cell plate will be 1x) into the alternating rows with (+) stimulation, and 20 μL of vehicle buffer for the alternating rows without (-) compound. Return the cell plate at 37 °C in 5% CO2 and incubate for at least 16 h.
4. Primary screening: Luminescence reading
- Prepare the Glo reagent, modified from Baker and Boyce21: 108 mM Tris–HCl; 42 mM Tris-Base, 75 mM NaCl, 3 mM MgCl2, 5 mM Dithiothrei-tol (DTT), 0.2 mM Coenzyme A, 0.14 mg/ml D-Luciferin, 1.1 mM ATP, 0.25% v/v Triton X-100, 2 mM Sodium hydrosulfite.
NOTE: Stock solutions of the reagents can be made in advance, except for D-Luciferin, which is always freshly added to the Glo reagent in its powdered form. If black plates were used, the amount of D-Luciferin can be increased up to 0.25 mg/mL. - At 16–24 h following stimulation, decant the transfected cell media by gently flicking the 384-well plate over the sink and taping it over a paper towel. Add 20 μL/well of Glo reagent and incubate the plate at room temperature for 5–20 min. Read the plates using a microplate luminescence counter, with an integration time of 1 s/well.
5. Primary screening: Data analysis
- Export the saved files from the luminescence counter as a spreadsheet; results will be recorded in relative luminescence units (RLU). Based on the layout of the 384-well plate, calculate the activation (fold change) of each receptor using the following formula:
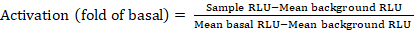
NOTE: Here Sample RLU refers to value of each of the four replicate wells of the stimulated (+ compound) quadrant, Mean background RLU is the mean of the negative controls on the plate, and Mean basal RLU refers to the mean of the untreated quadrant of that same receptor (- compound). In addition, calculate the standard deviation of the 4 data points to verify the quality of the results. It is recommended to perform a log2 transformation on the mean of the fold changes to rectify any heteroskedasticity; the log2 base is a practical choice to help identify positive hits. Empirically set the positive hit thresholds; it has to be noted that some receptors can have as low as 2-fold increase and up to 40-fold increase for others with full agonist. - Based on the results, select the GPCRs that are potential positive hits for secondary screening.
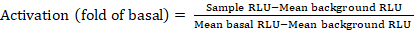
6. Secondary screening: Cell seeding and transfections
- Subculture HTLA cells in 100 mm dishes at a total cell density of 5 x 106 cells in 11 mL of complete media (4.55 x 105/mL) and incubate at 37 °C for 24 h. If a same-day transfection is preferred, seed cells at a density of 7.5 x 106 cells and perform the transfection 4 h later.
- Pre-warm the reagents needed for calcium phosphate precipitation at room temperature. Combine 450 μL of 0.1x TE buffer with 50 μL of 2.5 M CaCl2 and quickly vortex; these amounts are specific for one 100 mm dish, based on the volume of growth medium it holds.
- In a tube, add 500 μL of the TE/CaCl2 solution to 10 μg of GPCR cDNA and vortex. Add 500 µL of 2x Hepes buffer solution in the tube, shake vigorously (do not vortex), and incubate for 1 min.
NOTE: 1 µg of any plasmid encoding a fluorescent protein (e.g. YFP, mCherry, etc.) can be co-transfect with 9 µg of GPCR cDNA for a total of 10 μg. The fluorescent protein is used to track transfection efficiency, and this minimal amount will not interfere with the assay. - Immediately following the short incubation, dispense the 1 mL solution dropwise onto the cells. Gently rock the plate back and forth to evenly distribute the precipitate, taking care not to swirl the plate, and incubate at 37 °C for 24 h.
- The following day, observe the transfection efficiency by looking at the expression of the fluorescent protein under a fluorescent cell imager; transfections greater than 50% coverage are ideal.
- Incubate the necessary 384-well PLL-coated plate(s) in the incubator at 37 °C to warm it before seeding cells. Remove the storage solution of 1x antibiotic-antimycotic from the 384-well PLL-coated plate(s) by flicking the plate over the sink and taping it over a paper towel to dry.
- Gently rinse the transfected cells with Versene solution (1X PBS, pH 7.4; 0.53 mM EDTA), and detach by adding 3 mL of 0.05% trypsin/0.53 mM EDTA to the dish. Transfer the contents to a centrifuge tube containing at least an equal amount of complete DMEM to neutralize the trypsin.
- Spin down the cells at 500 x g for 3 min and resuspend the cells at a density of 0.4 x 106 cells/mL in starving media. Seed cells into the 384-well PLL-coated plate(s) at a final density of 25,000 cells/well by dispensing 45 μL of the cell suspension using an electronic multichannel pipet. Return the plates to the 37 °C for a minimum of 4 h, allowing the cells to properly attach to the wells before proceeding to the stimulation.
7. Secondary screening: Drug plate preparation for 16-point (half log) dose-curve
- In a 96-well plate, add 270 μL of 1X HBSS drug buffer (1x Hank's Balanced Salt Solution [HBSS], 20 mM HEPES pH 7.4, 1x antibiotic-antimycotic), excluding the last row (row H) of the plate, as shown in Figure 4.
NOTE: For peptides, colloidal molecules, and poorly water-soluble compounds, the addition of 0.1–1% BSA is suggested. To prevent drug oxidation, up to 0.01% ascorbic acid can also be added. - From the drug stock, prepare a drug solution (referred to as the "High" concentration) by calculating a final 3x concentration (final concentration of the drug in the cell plate will be 1x). As an example, for a dose–response curve with 10 μM as its highest concentration, prepare the "High" concentration at 30 μM. Pipet 300 μL of "High" concentration into wells in row H.
- In another tube, prepare the "Low" concentration, which represents the "High" concentration divided by 3.16 (half-log). Based on the previous example, the "Low" concentration would be 9.49 μM. Pipet 300 μL of "Low" concentration into wells in row H, adjacent to the "High" wells.
NOTE: The total number of 96-wells needed in row H will depend on the number of cells and stimulation conditions. Four wells (two "High" and two "Low") will have ample drug solution to stimulate an entire 384 well plate. - Perform a serial dilution by pipetting 30 μL of drug solution from the "High" and "Low" wells of row H to the previous row (row G) and mix by manually pipetting up and down, or as recommended, using an electronic multichannel pipette with the "Pipette and Mix" function. Repeat this step up until the first and most diluted row (row A), while discarding tips between dilutions.
NOTE: If desired, the serial dilutions can be stopped before row A, representing an internal control with no drug, in other words, a "true zero". - Using Figure 4 as a reference, stimulate transfected cells by pipetting 20 μL of the "Low" column dilutions from the 96-well plate to rows A–O of the previously seeded 384-well plate, as well as 20 μL of the "High" column dilutions to wells B–P. Incubate the plate at 37 °C for a minimum of 16 h.
8. Secondary screening: Luminescence reading and data analysis
- At 16–24 h following stimulation, decant the transfected cell media by gently flicking the 384-well plate over the sink and taping it over a paper towel. Add 20 μL/well of Glo reagent and incubate the plate at room temperature for 5–20 min. Read the plates using a microplate luminescence counter, with an integration time of 1 s/well.
- Export the saved files from the luminescence counter as a spreadsheet; results will be recorded in relative luminescence units (RLU). Transfer the data of the 384-well plate to a statistics software to analyze the results using its built-in XY analysis for non-linear regression curve fit. Select the built-in 3-parameter dose-response stimulation function "Log(agonist) vs. response (three parameters)",

NOTE: Here Top and Bottom are plateaus in the units of the Y axis, respectively the maximal response and basal level, EC50 is the concentration of the agonist that that generates 50% response between Top and Bottom, and X refers to the log concentration of the agonist. This model assumes that the dose-response curve has a standard Hill slope of 1.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Using the PRESTO-Tango protocol presented herein, a chromaffin granule (CG) extract was screened against 168 non-olfactory GPCR targets, with the majority being orphan receptors. Profiling of said extract was performed by examining β-arrestin2 mobilization at the chosen receptors, based on the principle designed by Barnea et al.18 (Figure 1). Plasmid cDNA of the GPCRs of interest was taken from the PRESTO-Tango GPCR Kit and assembled in two 96 well-plates in the desired layout. In total, four 384-well plates seeded with HTLA cells were transfected, as each half of the 96-well DNA plates was made into a full 384-well plate, resulting in each receptor being transfected in two quadrants (eight 384-wells total). One of the two quadrants was stimulated with the CG extract; put differently, alternating rows C–D, G–H, K–L, and O–P represented the (+) stimulation (Figure 2). Out of the 168 GPCRs that were interrogated in the primary screening, only two receptors were contenders as potential active targets, specifically dopamine receptor D3 (DRD3) and opsin 5 (OPN5). DRD3 produced a significant log2fold change of 4.70, whereas OPN5 produced a slightly lower response of 2.39, both meeting the threshold cut-off of log2 fold change >2. In comparison, the positive control for the primary screen was DRD2 stimulated with quinpirole, a selective agonist of this receptor, and produced a log2 fold change of 4.58 (Figure 3). To reproduce these signal windows and eliminate the possibility of false-positive hits, a secondary screen was conducted with the aforementioned receptors. Besides testing the CG extract, given that DRD3 is a non-orphan receptor, another condition was prepared as a positive control, specifically stimulation with quinpirole, one of its selective agonists. On the other hand, OPN5 is an orphan receptor and as such, no reference agonist can be tested alongside the CG extract as a positive control; only buffer was tested as a negative control. Further pharmacological characterization of these two GPCRs was performed by preparing 16-point dose curves ranging from 10-5 M to 10-12.5 M. Specifically, the CG extract and quinpirole stock solutions at 10 mM were diluted to 30 µM and 9.49 µM, the corresponding "High" and "Low" concentrations in bottom row (row H) of the 96-well drug plate; these formulations will become 10-5 M and 10-5.5 M once 20 µL is dispensed on to the transfected cells in 40 µL of starved medium, for a total of 60 µL within each 384-well. As previously described, serial dilutions were performed such that the most dilute drug formations for 10-12 M and 10-12.5 M are in the top row (row A) (Figure 4). Dose-response curves from the secondary screening were created using GraphPad Prism to evaluate ligand potency and efficacy. In comparison to quinpirole, the CG extract produced similar signal windows and EC50 values, confirming its validity as an active hit at DRD3. However, a flat dose-curve similar to the negative control was produced for OPN5, ruling it out as a possible target for the CG extract (Figure 5).

Figure 1: Modular design of TANGO constructs (A) and general scheme for the β-arrestin (Tango) recruitment assay (B). (A) The GPCR Tango constructs consist of various module elements in the following order: an HA signal/FLAG tag, the GPCR CDS, a Vasopressin receptor 2 C-terminal tail, TEV protease cleavage site, and a tTA transcription factor. (B) The principle of the Tango assay involves transiently transfecting the GPCR Tango plasmids in HTLA cells, HEK293T cells stably expressing a β-arrestin2-TEV protease fusion protein and a luciferase reporter gene whose expression is activated by tTA. Activation of the GPCR will eventually result in the mobilization of the β-arrestin2-TEV to the receptor, bringing the protease in close proximity to its cleavage site. As a result, the tTA is cleaved from the GPCR tail, freeing the transcription factor to translocate into the nucleus and activate luciferase expression. Please click here to view a larger version of this figure.

Figure 2: Layouts of 96-well cDNA plate and 384-well cell plate for transfection and stimulation in PRESTO-TANGO primary screening. Depicting the preparation of a 384-well cDNA source plate for transfection, GPCR Tango constructs are first transferred from one half of a 96-well plate into a full 384-well plate, with each receptor being transfected in octuplicate. In this setting, stimulation of cells with (+) and without (-) the drug(s) of interest will occur in quadruplicate for each individual receptor. Please click here to view a larger version of this figure.

Figure 3: Graphical representations of hit identification from PRESTO-Tango primary screening. As a proof-of-concept, the biological activity of a chromaffin granule (CG) extract on the GPCRome was analyzed. HTLA cells were transfected in 384 well plates with 168 GPCR Tango constructs, and either stimulated with the CG extract (+ compound) or with vehicle buffer (- compound). pcDNA3.1 was used as a negative control, and DRD2 receptor stimulated with quinpirole was used as a positive control. The signal windows (A) and the log2 fold change (B) in receptor activation was calculated between the wells in the absence or presence of CG extract. All error bars represent SD (n = four measurements). Please click here to view a larger version of this figure.

Figure 4: Layout of 96-well drug plate preparation for stimulation in secondary screening. Depicting the preparation of a 96-well drug plate for cell stimulation, serial dilutions for a 16-point dose curve range start at 10-5 M (final concentration) in row H, with half-log intervals between each point until 10-12.5 M in row A. "High" and "Low" drug columns are used to stimulate alternating rows of the seeded 384-well plate. Please click here to view a larger version of this figure.

Figure 5: Dose-curve responses for compound profiling and demonstration of β-arrestin2 recruitment to GPCRs in secondary screening. HTLA cells were transiently transfected with receptors DRD3 (A) and OPN5 (B). Both transfected conditions were stimulated with a CG extract in half-log increments, as well as the DRD3 specific agonist quinpirole as a positive control, and vehicle buffer for OPN5 as a negative control. All error bars represent SD (n = three measurements). Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
The conformationally dynamic GPCRs are powerhouses of signal transduction. The physiochemical properties of the binding pockets of these heptahelical receptors, as well as their physiological relevance underscore the need for GPCR ligand screening tools. As presented above, the PRESTO-Tango assay is rapid, sensitive and user-friendly, lending itself to drug development. Not only does this assay measure agonist-induced activation, but it can also be used to quantify the activity of antagonists and allosteric modulators19. In light of functional selectivity, a concept which suggests that different drug structures can elicit different receptor signaling cascades at a single receptor, comparing the activation of the G-protein pathway using G-protein dependent assays with β-arrestin recruitment using PRESTO-Tango could provide cues for the designing lead compounds with reduced negative side effects. Notably, its independence from detecting G-protein coupling helps identify coupling partners for orphan GPCRs that would not be have been previously detected by G-protein dependent assays.
To ensure consistency and robustness of PRESTO-Tango screens, care must be taken in all steps of the protocol, as perturbations introduced will be magnified due to the nature of this platform. Of course, there are general measures common to all HTS screens which should be taken into consideration, such as using reagents of the same lot/formulation to ensure identical stability and biological activity throughout, as well as keeping conditions on the HTS system consistent such as cell seeding density and drug incubation time. The miniaturized format of PRESTO-Tango demands attention to a couple of critical points: variation in cell seeding density and their homogeneous distribution (clumping versus single cell suspension) between wells, low transfection efficiency, and poor compound stimulation and delivery will prevent day-to-day and plate-to-plate reproducibility. To that effect, triturate the HTLA cell suspension to homogenize the solution before seeding and ensure a 50–70% cell confluency before transfection. The vehicle for the delivery of the compounds should be verified, with dimethyl sulfoxide being the most common carrier. Typically, the highest concentration of our dose curves is 10 μM, but this may change depending on the nature and potency of the compound; it is important to test various concentrations to deduce cellular tolerance and toxicity.
Given that some GPCRs have high constitutive activity, one issue that may arise during screening is a reduced dynamic range and a background signal that is higher than expected. This can be somewhat mitigated by ensuring that serum starvation is being performed with DMEM medium supplemented with 1% dFBS. It should be taken into consideration that if the luminescence output is high enough, there can still be bleed-through into adjacent wells, which may result in erroneously calculated fold changes. Undetectable or low signals (assuming a response is expected) can be explained in a number of ways, namely poor expression of GPCR(s) in HTLA cells, the biological activity of the compound is lost rendering it inefficacious, or the receptor(s) in question do not intrinsically recruit β-arrestin2. Respectively, assessing the quantity and quality of transfected plasmid receptor DNA, testing other preparations/lots of the inefficacious compound in question, and performing orthologous protein-protein interaction techniques such as BRET/FRET or co-immunoprecipitation are some suggested solutions to this problem. In addition, receptor expression could also be improved by subcloning Tango receptor(s) of interest into lentiviral vectors and transducing HTLA cells, generating a HTLA-GPCR stable cell line. A shift in the expected potency of an agonist during secondary screening could imply that the drug stimulation time and/or concentration of compound needed to stimulate a response is insufficient, or that the drug plate serial dilutions were incorrectly prepared. Use of an electronic multichannel pipette or an automated pipettor system without changing tips in between when creating drug serial dilutions could be an issue when working with sticky compounds.
Notable differences between the original Tango assay developed by Barnea et al.18 and the PRESTO-Tango platform include the design of the receptor in a modular format, consisting of codon-optimized sequences, which improves receptor expression in mammalian cells, epitope tags to validate said expression, and restriction sites which flank GPCRs, V2 tail and TEVcs-tTA, enabling for excision of parts and subcloning. Most importantly, PRESTO-Tango surpasses the Tango assay in terms of screening power and experimental design. Quadruplicate sample testing of approximately 300 GPCRs is accomplished in only 8 384-well plates, while accounting for negative background controls and positive controls to monitor transfection efficiency. While PRESTO-Tango is suitable for screening the GPCR-ome with only one compound of interest, interrogation with multiple ligands can also be performed, albeit at increased cost and use of resources, such as with pooled or arrayed small molecule compound libraries or biological samples which consist of mixtures of various chemical entities. Granted, this issue can be mitigated by reducing the number of compounds to interrogate by performing chemical similarity and diversity analyses of the compound libraries in question. While the PRESTO-Tango platform is more applicable for primary screening purposes, secondary profiling can be performed at a smaller scale, in medium or low-throughput formats, to confirm the functional consequences of ligand stimulation. However, as with all other GPCR assays, it must be acknowledged that there are no suitable positive controls for orphan receptors during secondary screening with the Tango assay. Nonetheless, potential positive hits can be identified if the output data can be fitted to a sigmoidal dose-response curve, with a computed signal window and EC50 value. It is also important to note that the mechanism of ligand activity, be it for orphan or non-orphan receptors, cannot be elucidated without running parallel assays.
With all components of PRESTO-Tango already optimized, including HTLA cell line and GPCR Tango constructs, little room for modification is required apart from choice of compound formulation(s) to be used for drug stimulation. If desired, an HTLA cell line stably expressing a receptor can be easily generated by cloning said GPCR-Tango receptor within the recommended pIRESbleo3 vector (Clonetech), and selecting clones using zeocin. With regard to the swap from pcDNA3.1 to pIRESbleo3, simply digest the GPCR Tango construct with NotI and XbaI and insert into the destination vector at restriction sites NotI and NheI. Notwithstanding, there are avenues for adapting and optimizing this technology. One of the pillars of this technology are HTLA cells, a HEK293T cell line stably expressing a β-arrestin2-TEV fusion gene and a tTA-dependent luciferase reporter, graciously supplied from the lab of Richard Axel. While a crucial component of PRESTO-Tango, there are currently no other alternatives in terms of cell line origin, or the genes they express. Moreover, future engineered cell lines can be generated to express other TEV fusion genes to track other proteins besides β-arrestin2, specifically those that have been previously shown to interact or found in residence to GPCRs, such as 14-3-322, SAP9723, and β-arrestin1, which is the more prevalent isoform of non-visual arrestins in vertebrates24. This can be achieved by using the parental HTL cells that solely contained the luciferase reporter controlled by the tetO7 promoter. One limitation to PRESTO-Tango is non-specific activation of the reporter promoter. Based on a tetracycline-dependent regulatory system (tet system), the tetracycline-responsive element (TRE) controls expression of the downstream luciferase reporter. However, previous studies have demonstrated "leaky" expression of luciferase due to endogenous transcription factors25,26. As a result, some compounds could activate the reporter independently of the β-arrestin2 recruitment or GPCR activation, increasing the number of false positives. Another issue that emerges, also common to other HTS methods, are "frequent hitters", promiscuous compounds that stimulate substantial responses in several targets27. Nonetheless, the PRESTO-Tango's parallel screening set-up facilitates identification of these artifacts, which can be further tested to confirm their effect on luciferase activity. Altogether, PRESTO-Tango has provided solid foundations for the study of arrestin recruitment to GPCRs, and in the larger scheme of drug discovery, as a utile GPCR ligand screening and deorphanization tool.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authours declare no competing interests.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (CIHR grant #MOP142219).
Materials
| Name | Company | Catalog Number | Comments |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, BLACK, with lid, Sterile | NUNC | 12-566 | |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, White, with lid, Sterile | NUNC | 12-566-1 | |
| 384 Well Round Bottom, Polypropylene, Non-Treated, Blue, non-sterile, without lid | ThermoFisher | 12-565-390 | |
| Antibiotic-Antimycotic | Wisent | 450-115-EL | |
| D-Luciferin, sodium salt | GoldBio | LUCNA | |
| DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10-013-CV | |
| Eppendorf Xplorer, 12-channel, variable, 15–300 µL | Eppendorf | 4861000155 | |
| Eppendorf Xplorer, 12-channel, variable, 5–100 µL | Eppendorf | 4861000139 | |
| Matrix Platemate 2x3 | ThermoFisher | 801-10001 | |
| MicroBeta 1450 Wallac | Perkin Elmer | ||
| Penicilin-Streptomycin | Wisent | 450-201-EL | |
| Poly-L-Lysine hydrobromide | Millipore-Sigma | P2636-500MG | |
| Roth Lab PRESTO-Tango GPCR Kit | Addgene | Kit #1000000068 |
References
- Liapakis, G., et al. The G-protein coupled receptor family: actors with many faces. Current pharmaceutical design. 18 (2), 175-185 (2012).
- Pierce, K. L., Premont, R. T., Lefkowitz, R. J. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 3 (9), 639-650 (2002).
- Kroeze, W. K., Sheffler, D. J., Roth, B. L. G-protein-coupled receptors at a glance. Journal of cell science. 116, Pt 24 4867-4869 (2003).
- Rask-Andersen, M., Almén, M. S., Schiöth, H. B. Trends in the exploitation of novel drug targets. Nature Reviews Drug Discovery. 10 (8), 579-590 (2011).
- Ngo, T., et al. Identifying ligands at orphan GPCRs: current status using structure-based approaches. British journal of pharmacology. 173 (20), 2934-2951 (2016).
- Zhang, R., Xie, X. Tools for GPCR drug discovery. Acta pharmacologica Sinica. 33 (3), 372-384 (2012).
- Kimple, A. J., Bosch, D. E., Giguere, P. M., Siderovski, D. P. Regulators of G-Protein Signaling and Their G Substrates: Promises and Challenges in Their Use as Drug Discovery Targets. Pharmacological Reviews. 63 (3), 728-749 (2011).
- Wettschureck, N., Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiological Reviews. 85 (4), 1159-1204 (2005).
- Denis, C., Saulière, A., Galandrin, S., Sénard, J. -M., Galés, C. Probing heterotrimeric G protein activation: applications to biased ligands. Current Pharmaceutical Design. 18 (2), 128-144 (2012).
- Yin, H., et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. The Journal of Biological Chemistry. 284 (18), 12328-12338 (2009).
- Cheng, Z., et al. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Current Chemical Genomics. 4, 84-91 (2010).
- Roth, B. L., Kroeze, W. K. Integrated Approaches for Genome-wide Interrogation of the Druggable Non-olfactory G Protein-coupled Receptor Superfamily. The Journal of Biological Chemistry. 290 (32), 19471-19477 (2015).
- Jean-Charles, P. -Y., Kaur, S., Shenoy, S. K. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. Journal of Cardiovascular Pharmacology. 70 (3), 142-158 (2017).
- Smith, J. S., Rajagopal, S. The β-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of Biological Chemistry. 291 (17), 8969-8977 (2016).
- Böhme, I., Beck-Sickinger, A. G. Illuminating the life of GPCRs. Cell Communication and Signaling. 7 (1), 16 (2009).
- Fang, Y. Label-Free Receptor Assays. Drug discovery today. Technologies. 7 (1), 5-11 (2011).
- Wang, T., et al. Measurement of β-Arrestin Recruitment for GPCR Targets. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences. , (2004).
- Barnea, G., et al. The genetic design of signaling cascades to record receptor activation. Proceedings of the National Academy of Sciences of the United States of America. 105 (1), 64-69 (2008).
- Kroeze, W. K., et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nature Structural & Molecular Biology. 22 (5), 362-369 (2015).
- Jordan, M., Schallhorn, A., Wurm, F. M. Transfecting Mammalian Cells: Optimization of Critical Parameters Affecting Calcium-Phosphate Precipitate Formation. Nucleic Acids Research. 24 (4), 596-601 (1996).
- Baker, J. M., Boyce, F. M. High-throughput functional screening using a homemade dual-glow luciferase assay. Journal of Visualized Experiments. (88), 50282 (2014).
- Li, H., Eishingdrelo, A., Kongsamut, S., Eishingdrelo, H. G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal Transduction and Targeted Therapy. 1 (1), 16018 (2016).
- Walther, C., Ferguson, S. S. G. Minireview: Role of intracellular scaffolding proteins in the regulation of endocrine G protein-coupled receptor signaling. Molecular Endocrinology. 29 (6), Baltimore, Md. 814-830 (2015).
- Gurevich, E. V., Benovic, J. L., Gurevich, V. V. Arrestin2 expression selectively increases during neural differentiation. Journal of Neurochemistry. 91 (6), 1404-1416 (2004).
- Shaikh, S., Nicholson, L. F. B. Optimization of the Tet-On system for inducible expression of RAGE. Journal of Biomolecular Techniques JBT. 17 (4), 283-292 (2006).
- Blau, H. M., Rossi, F. M. Tet B or not tet B: advances in tetracycline-inducible gene expression. Proceedings of the National Academy of Sciences of the United States of America. 96 (3), 797-799 (1999).
- Roche, O., et al. Development of a Virtual Screening Method for Identification of "Frequent Hitters" in Compound Libraries. Journal of Medicinal Chemistry. 45 (1), 137-142 (2002).
