

Summary
Viene descritto un metodo altamente parallelo per misurare la scissione specifica del sito del DNA a livello di singola molecola. Questo protocollo illustra la tecnica utilizzando la restrizione endonuclease NdeI. Il metodo può essere facilmente modificato per studiare qualsiasi processo che si traduce in scissione del DNA specifica del sito.
Abstract
La scissione del DNA specifica del sito (SSDC) è un passo chiave in molti processi cellulari ed è fondamentale per l'editing genico. Questo lavoro descrive un saggio cinetico in grado di misurare SSDC in molte singole molecole di DNA contemporaneamente. I DNA del substrato con legato al tallone, ciascuno contenente una singola copia della sequenza di destinazione, vengono preparati in un canale di flusso microfluidico. Un magnete esterno applica una forza debole alle perline paramagnetiche. L'integrità di un massimo di 1.000 singoli DN può essere monitorata visualizzando le microsfere sotto l'imaging darkfield utilizzando un obiettivo di ingrandimento ampio e basso campo. L'iniezione di una restrizione endonucleasi, NdeI, avvia la reazione scissione. La microscopia video viene utilizzata per registrare il momento esatto di ogni scissione del DNA osservando il fotogramma in cui il tallone associato si muove verso l'alto e verso l'alto dal piano focale dell'obiettivo. Il conteggio del tallone fotogramma per fotogramma quantifica la reazione e una adattamento esponenziale determina il tasso di reazione. Questo metodo consente la raccolta di dati quantitativi e statisticamente significativi sulle reazioni SSDC a singola molecola in un singolo esperimento.
Introduction
La scissione del DNA specifica del sito (SSDC) è un passo chiave in molte transazioni genomiche. Ad esempio, i sistemi di restrizione batterica (RM)1 e CRISPR2 proteggono le cellule dagli attacchi di fagi e plasmi, riconoscendo e fendendo il DNA estraneo in sequenze specifiche. Nel tipo II RM, le endonucleasi di restrizione (RSI) riconoscono brevi sequenze di coppie di base (bp) 4-8 tramite interazioni proteina-nucleicodell'acido 3. Le endonucleasi associate a CRISPR, come Cas9, si legano ai siti tramite ibridazione del sito di destinazione con crRNA associati alle endonucleasi4. La creazione di DSB (Double Stranded Breaks) specifiche del sito sono anche il primo passo in molti eventi di ricombinazione del DNA5. Ad esempio, la diversità delle regioni di associazione dell'antigene create dalla ricombinazione V(D)J richiede il riconoscimento e la scissione di siti di destinazione specifici6. Alcuni trasposoni sono noti per indirizzare sequenze di DNA specifiche, cosìcome 7. Non sorprende che molte nucleasi specifiche del sito coinvolte in questi processi, come Cas9, siano una componente chiave delle tecnologie di editing genico8. Inoltre, sono state progettate nuove endonucleasi specifiche del sito (ad esempio, nucleasi di dita di zinco9 e TALENS10) per modificare i genomi.
Molti metodi sono stati impiegati per misurare la cinetica della scissione site-specific degli acidi nucleici. Questi includono l'analisi del gel, la fluorescenza11,12e i metodi basati sul sequenziamento13. Un importante progresso è stato raggiunto con il tethering delle microsfere, che consente ai DSB in singole molecole di DNA di essere rilevati dal movimento di una perla dopo la separazione del filamento. In questi metodi, diversi tipi di forze sono impiegati per garantire la separazione del filo e il movimento del tallone post-scissione. In un caso, sono state utilizzate trappole ottiche per misurare la scissione del DNA da parte di EcoRV14. In questi esperimenti, la ricerca target è l'obiettivo dell'indagine, con condizioni ottimizzate in modo che l'associazione specifica del sito sia il passaggio di limitazione della frequenza. Uno svantaggio delle trappole ottiche è che si può osservare un solo DNA alla volta. Inoltre, è necessario applicare una forza di trazione di grandi dimensioni periodica per testare la separazione del filo.
Un'altra tecnica utilizza una combinazione di flusso e deboli forze magnetiche per tirare sul tallone in modo continuo15. In questo modo, viene misurata la scissione limitata di NdeI. Il metodo impiegato consente la misurazione simultanea di diverse centinaia di DNA contemporaneamente, consentendo di raggiungere la significatività statistica in un singolo esperimento. Sono stati utilizzati anche esperimenti basati su pinzette magnetiche. In uno di questi studi, un integratore retrovirale è stato studiato includendo un DSB nell'oligonucleotidedi inserimento 16. Il successo dell'integrazione ha portato all'incorporazione di DSB nel DNA legato e alla perdita del tallone allegato. In uno studio simile sulla restrizione endonuclea EcoPI, dipendente dall'ATP, sono state osservate decine di DNA in un singolo esperimento17. Le pinzette magnetiche detengono il vantaggio che la tensione, così come il looping del DNA, possono essere controllate e monitorate durante la reazione.
Qui è presentato un metodo a singola molecola altamente parallelo per misurare la cinetica SSDC, che sfrutta i recenti miglioramenti nel tethering su larga scala dei DNA. Questo metodo è un miglioramento e l'estensione dei metodi precedenti utilizzati per misurare la replicazione del DNA18, lunghezza del contorno del DNA19e scissione da Es15. In questa tecnica, i DNA lineari contenenti una singola copia della sequenza di riconoscimento vengono preparati con la biotina ad un'estremità e la digoxigenina all'altra. La biotina lega streptavidin, che è covalentemente ad una microsfera paramagnetica. I complessi di perline di DNA vengono iniettati in un canale microfluidico che è stato funzionalizzato con frammenti DI FAB anti-digoxigenina. Il DNA si lega quindi ai punti di attacco della superficie attraverso il legame della digoxigenina ai frammenti FAB adsorbti. Le deboli forze magnetiche applicate con un magnete permanente impedise al tallone di attaccarsi in modo non specifico alla superficie. I campioni possono essere iniettati rapidamente (<30 s) nel canale di flusso per attivare la reazione di scissione. Il flusso viene disattivato durante la raccolta dei dati. Come ogni DNA è fessurato, il tempo esatto di scissione può essere determinato registrando il fotogramma in cui il tallone si muove su e fuori dal piano focale dell'obiettivo, scomparendo così dalla registrazione video. Un conteggio fotogramma per fotogramma delle perline rimanenti può essere utilizzato per quantificare il progresso della reazione.
Di seguito è riportato il protocollo completo e i dati di esempio raccolti utilizzando NdeI. Come esempio di come la tecnica può essere applicata, i tassi di scissione per una gamma di concentrazioni proteiche sono misurati a due diverse concentrazioni di magnesio, un cofattore di metallo essenziale. Anche se questa applicazione del protocollo utilizza NdeI, il metodo può essere adattato per l'uso con qualsiasi nucleasi specifica del sito variando il design del substrato di DNA.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Fare la cella di flusso
- Lavaggio delle coperture
- Mettere i coverlips in vasetti di colorazione e sonicare con etanolo (EtOH), quindi con 1 M KOH (per 30 min ciascuno). Per evitare precipitazioni KOH in EtOH, risciacquare accuratamente con ddH2O tra lavaggi.
- Ripetere entrambi i passaggi di lavaggio EtOH e KOH 1x per un numero totale di quattro lavaggi (due EtOH e due KOH). Conservare i coperture pulite in ddH2O in vasetti di colorazione.
- Tagliare i tubi di carico e di uscita (8 cm di lunghezza) utilizzando un rasoio pulito e inserire nei fori in un vetrerà di vetro pulito. Utilizzare PE-20 per l'ingresso e PE-60 per l'uscita. Epossiy per 5 minuti per fissare tubi e tagliare qualsiasi tubo in eccesso.
NOTA: Vetrini di vetro misura 2'' x 3'' x 1 mm. I fori sono forati a coppie di 15 mm di distanza. Ogni coppia forma entrambe le estremità di un singolo canale. Il tubo con l'ID più piccolo viene utilizzato per l'ingresso, in quanto ciò riduce il volume morto a monte e quindi il tempo di miscelazione richiesto (Figura 1). - Allineate e applicate il nastro pretagliato a due lati con il motivo del canale tagliato sopra i fori sul vetro di vetro. Affiante con le forcepi di plastica per ottenere una buona tenuta.
NOTA: Il nastro a due lati utilizzato in questo esperimento è spesso 120 m e i canali sono 2 mm di larghezza e 15 mm di lunghezza. I canali vengono tagliati utilizzando una stampante a coltello (Tabella dei materiali). È possibile montare fino a quattro canali su un singolo coverlip. Vedere Figura 1 per un'immagine della cella di flusso. - Dopo aver staccato il supporto, applicare un coperchio pulito (essiccato con aria compressa) sul nastro e lisciare di nuovo con le foci di plastica per un buon sigillo.
- Epossiy il bordo fuori il coverslip per sigillare la cellula di flusso e lasciarlo curare.
2. Preparazione del DNA etichettato per il tethering
- In un tubo PCR, preparate 50 L del mix di reazione PCR contenente la polimerasi del DNA ad alta fedeltà 0,02 U/L, 200 dNTP M, primer in avanti da 0,5 M, primer inverso da 0,5 M e 250 ng di DNA vettoriale M13mp18.
NOTA: Qui, il primer in avanti (biotina-CCAACTTAATCGCCTTGC) e il primer inverso (digoxigenin-TGACCATTAGATACATTTCGC) sono stati scelti per amplificare una regione lunga circa 1.000 bp, che va dalle posizioni 6338 a 107 nel genoma circolare del DNA M13mp18. C'è un unico sito NdeI nel mezzo della regione amplificata. Il primer in avanti 5ʹ etichettato con digoxigenina, che lega l'anti-digoxigenina sul coperture. Il primer inverso 5ʹ etichettato con biotina, che lega le perline rivestite di streptavidi. - Inserite il tubo PCR nel termociclo e seguite il ciclo come mostrato nella tabella 1.
- Purificare il prodotto PCR con un kit di pulizia PCR seguendo il protocollo del produttore.
NOTA: Utilizzando il kit specificato nella Tabella dei Materiali,la resa tipica del DNA è di 2 g.
3. Tethering di DNA e perline
- Preparare 10 mL di buffer A (1 M Tris-HCl [pH - 7,5], 50 mM NaCl, 2 mM MgCl2, 1 mg/mL β-Casein, 1 mg/mL Pluronic F-127). Degas in un desiccatore sottovuoto per almeno 1 h.
- Per funzionalizzare la cella di flusso, iniettare 25 L di frammenti FAB anti-digoxigenina (20 g/mL) in PBS nel canale di flusso. Utilizzare punte di caricamento gel per adattarsi al tubo PE-60. Incubare a temperatura ambiente (RT) per 30 min.
- Dopo l'incubazione, svuotare il canale tirando 0,5 mL di buffer A attraverso il canale utilizzando una siringa. Fare attenzione a non introdurre aria nel canale.
- Dopo la funzionalizzazione, montare la cella di flusso su un microscopio invertito. Collegare il tubo di uscita a una pompa di siringa e mettere il tubo di ingresso in un tubo di microcentrifuge contenente tampone A.
- Tirare manualmente almeno 0,5 mL del buffer A per svuotare il sistema e far uscire la pompa. Lasciare che la pompa funzioni a 10 L/min per almeno 5 min per equilibrare il sistema.
- Per preparare le perline(Tabella dei materiali), vortice la bottiglia di brodo di perline e pipette 1,6 L delle perline di 10 mg/mL in 50 L del buffer A, quindi vortice.
- Utilizzando un separatore magnetico, pipette fuori il buffer e risuostare in 50 L del buffer A, quindi vortice.
- Ripetere il passaggio 3.7 2x per un totale di tre lavaggi. Per l'ultimo lavaggio, rispendere in 100 L di buffer A e vortice per ottenere una concentrazione finale di 160 g/mL.
- Per complessore il DNA e le perline, preparare prima 480 L di 0,5 pM etichettato dna substrato nel buffer A. Quindi, pipetta in 20 l di sospensione perline da 160 g/mL, assicurandosi di vortice le perline prima di pipettare. Posizionare su un rotatore per 3 min.
- Dopo 3 minuti, caricare immediatamente nel canale ad una velocità di flusso di 10 L/min per 15 min o fino a quando non si osserva un sufficiente tethering del tallone.
NOTA: Le perline non devono essere così densamente imballate da interagire tra loro sulla superficie (vedere la sezione discussione). - Per lavare il canale di tutte le perline libere, impostare il tubo di ingresso su un tubo fresco di tampone A e fluire a 50 L/min per almeno 10 min o fino a quando non si osservano perline sciolte.
4. Raccolta e analisi dei dati
- Per preparare la raccolta dei dati, inserire il tubo di ingresso in un tubo di microcentrifuge contenente almeno 100 L di NdeI (0,25–4,00 U/mL) nel buffer A. Abbassare il magnete permanente sul canale di flusso e posizionare la sorgente luminosa fuori asse per l'imaging darkfield.
NOTA: Due magneti anulari di terre rare, epossidati insieme, sono tenuti 8 mm sopra la superficie attiva del canale di flusso utilizzando un post ottico a sbalzo durante la raccolta dei dati. Una lampada a collo d'oca viene utilizzata per il corso di luce fuori asse. - Utilizzare un microscopio commerciale, una videocamera e un software di raccolta dati(Tabella dei materiali). Nel software, fare clic sulla scheda "Esposizione" e impostare "Tempo esposizione" su 10 ms. Fare clicsulla scheda " Timelapse" e impostare "Numero immagini" su 600, "Durata" su 20 min e "Intervallo" su 2 s. Fare clic su" Esegui" per avviare la raccolta dei dati.
- Su una pompa di siringa, impostare la velocità di flusso su 150 L/min e il volume di iniezione su 80 l. Premere "Esegui" a 1 min nella raccolta dei dati. Dopo l'iniezione, spegnere la pompa e chiudere la valvola per evitare il flusso durante la raccolta dei dati.
- Una volta raccolti i dati, aprire il software di analisi delle immagini (Tabella dei materiali). Nella scheda "File", scegliere" Importa " "Sequenza immagine". Individuare i file di immagine nel menu a comparsa e fare clic su "Apri".
- Impostare la soglia scegliendo "Regola la proprietà" nel menu a discesa " Immagine ".Image Utilizzare la barra di scorrimento per impostare il valore di soglia per identificare i punti luminosi corrispondenti alle perline nell'immagine.
- Contare i punti luminosi in ogni fotogramma facendo clic su "Analizzaparticelle "nelmenu a discesa " Analizza ". Fare clic su" OK", quindi scegliere "Sì" per elaborare tutte le immagini. Salvare il file dei risultati.
NOTA: questo salverà un file di dati che contiene il numero di perline in ogni fotogramma video registrato. - Aprire il software di analisi dei dati (Tabella dei materiali) e importare il file dei risultati facendo clic su " Importa da file ditesto" nel menu a discesa "File". Tracciare i dati del conteggio delle perline rispetto al tempo.
- Adattare il numero di perline rispetto al tempo facendo clic su "Adattacurva "nelmenu a discesa " Analizza ". Sceglierel'equazione " EsponenteNaturale " e fare clic su "Prova adatta " "OK".
NOTA: solo i dati registrati dopo l'iniezione del campione devono essere inclusi nella regione di raccordo. Il parametro fit nell'esponente della funzione di raccordo sarà la velocità di scissione.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Utilizzando questa tecnica, i tassi SSDC di NdeI sono stati misurati per una gamma di concentrazioni proteiche (0,25–4,00 U/mL) a due diverse concentrazioni di magnesio (2 mM e 4 mM). Ognuna di queste condizioni è stata replicata almeno due volte, con poche centinaia di 1.000 DNA con tethering per esperimento. Figura 2 descrive la progettazione sperimentale. Nella figura 3 sono illustrati esempi di dettagli di raccolta e analisi dei dati. La figura 4 illustra come il tasso dipenda dalla concentrazione di proteine alle due concentrazioni di magnesio. Si può osservare che a concentrazioni proteiche sufficientemente basse, il tasso è proporzionale alle proteine e indipendente dal magnesio. Per concentrazioni proteiche sufficientemente elevate, il tasso dipende dal magnesio ma indipendente dalla concentrazione proteica.
| Passo | Descrizione | Temperatura (C) | Tempo (s) |
| 1 | Denaturazione | 98 | 30 |
| 2 | Sciogliere | 98 | 10 |
| 3 | Anneal | 60 | 30 |
| 4 | Estendere | 72 | 30 |
| 5 | Estendi estendi finale | 72 | 120 |
Tabella 1: parametri PCR. Sono mostrate le temperature e le durate dei passaggi del programma termociclo utilizzati nel passaggio 2.2 del protocollo. I passaggi di fusione, anneal ed estensione (passaggi 2, 3 e 4) vengono ripetuti 30x.
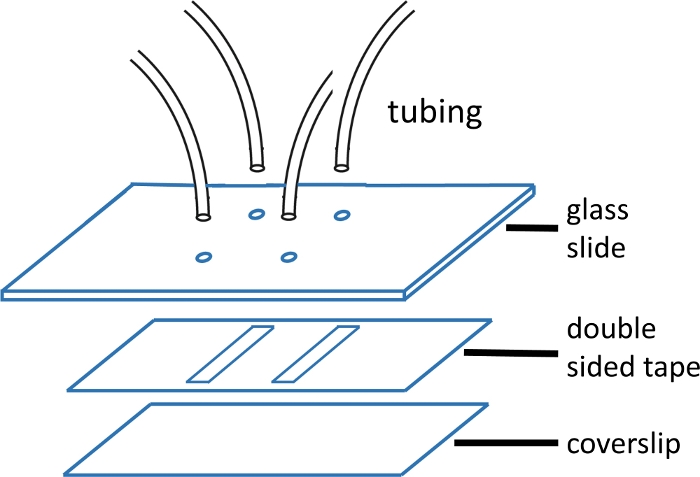
Figura 1: costruzione di celle di flusso microfluidiche.
Lo scivolo in vetro superiore (2'' x 3'', 1 mm di spessore) è pre-forato con fori che corrispondono al modello di canale. I tubi di ingresso e di uscita vengono inseriti nei fori e fissati con epossidia prima di attaccare il nastro adesivo e il vetro di copertura. Il nastro a due lati è pre-tagliato con motivo di canale. La diapositiva inferiore (#1 o #1.5) viene precedentemente pulita utilizzando il protocollo descritto nel testo principale. Una volta assemblato, il bordo del vetro di copertura è sigillato con epossiccia. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.

Figura 2: Progettazione sperimentale.
(A) Metodo di tethering del DNA. Il DNA a doppio filamento (1 kbp) etichettato con digoxigenina sull'estremità 5ʹ è attaccato alla superficie della cellula di flusso tramite interazione antidigoxigenina-digoxigenina. L 3ʹ fine del DNA, etichettato con biotina, è attaccato a una microsfera tramite interazione streptavidin-biotina. Il sito di scissione NdeI si trova al centro del DNA. (B) Configurazione sperimentale durante la raccolta dei dati che mostra la posizione del magnete e dell'obiettivo. Il magnete permanente mantiene una forza verso l'alto debole sul tallone durante la reazione di scissione. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
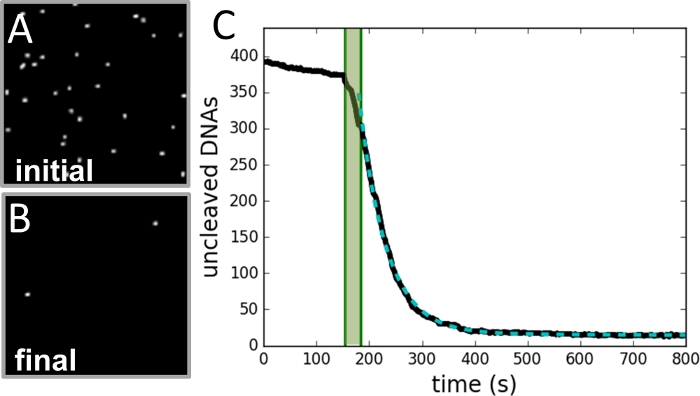
Figura 3: Esempio di raccolta e analisi dei dati.
(A) Immagine della regione di perline prese prima che venga avviata la reazione di scissione. (B) Immagine della stessa regione al termine della reazione. (C) Stampa del numero di perline rispetto al tempo (curva nera) come determinato da ogni fotogramma della registrazione video. L'area verde ombreggiata segna il periodo di iniezione di enzima e non è inclusa nella vestibilità. I dati si adattano a una singola curva esponenziale (curva verde tratteggiata), con una costante di decadimento uguale alla velocità di reazione. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.

Figura 4: La scissione NdeI dipende dalle concentrazioni di proteine e magnesio.
Grafico del tasso misurato di SSDC di NdeI per una gamma di concentrazioni proteiche a due diverse concentrazioni di magnesio: 2 mM (cerchi blu) e 4 mM (quadrati verdi). Le barre di errore rappresentano SEM. Le curve tratteggiate sono linee di tendenza e non rappresentano adattamenti alla teoria. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Il protocollo può essere utilizzato per misurare la cinetica di qualsiasi sistema SSDC, a condizione che la separazione del filo sia osservata durante l'esperimento. La rilevazione della scissione è influenzata osservando il distacco del tallone legato e quindi segna l'istante di separazione del filo. Tutti i passaggi precedenti si verificano prima del rilevamento della scissione; pertanto, viene registrato solo il tempo di transito totale.
Il cosorpecco cellulare di flusso è funzionalizzato tramite l'adorption non specifico della proteina anticorpale al vetro pulito. Il vetro pulito in modo insufficiente può influenzare il legame dell'anticorpo. Nel tethering, la densità del tallone dovrebbe essere sufficientemente bassa in modo che le perline non interagiscano. La densità superficiale dei punti di attacco può essere controllata dalla concentrazione di anticorpi durante la funzionalizzazione. Il numero totale di perline dipende dalle dimensioni del campo di visualizzazione. In questo caso, alcune centinaia a 1.000 perline con tethered erano sufficienti per buone statistiche ed evitavano interazioni perline. Durante l'iniezione di perline, il tethering superficiale è stato monitorato tramite video in diretta. L'iniezione di perline è stata interrotta quando il numero stimato di perline era tra 500 e 1.000 perline.
I tassi di scissione più veloci che possono essere misurati con precisione sono limitati dal tempo di miscelazione della cella di flusso. Il tempo di miscelazione nelle cellule di flusso laminare è influenzato da diversi fattori. La diffusione in superficie è un passo chiave; pertanto, il tempo di miscelazione dipende dal coefficiente di diffusione del reante. La cesoia significativa che si verifica nel tubo di ingresso, che trasporta il campione al canale di flusso dal serbatoio del campione, può aumentare il tempo necessario per garantire un'adeguata miscelazione alla superficie di reazione nel canale. Si è scoperto che il tempo di miscelazione potrebbe essere ridotto riducendo il volume morto a monte e aumentando la velocità di flusso. Con un diametro interno di 380 m e una lunghezza massima di 8 cm per il tubo di ingresso (e la velocità di flusso di 150 l/min), si è scoperto che il tempo di iniezione poteva essere ridotto a 20 s senza influire sulla velocità di scissione misurata. Poiché il tempo di miscelazione dipende dal coefficiente di diffusione del reagonfio, deve essere determinato separatamente per ogni enzima o attivatore di scissione studiato.
Il metodo di tethering consente la rottura del tethering non specifica, presumibilmente a causa della dissociazione del complesso di digoxigenina-anticorpo o del rilascio dell'anticorpo dalla superficie. Ciò si traduce in un tasso di perdita di perline di fondo riproducibile presente prima dell'iniezione di enzima di 3 x 10-4 s-1. Questo effetto sistematico può essere corretto sia sottraendo la velocità di fondo dalla velocità di scissione misurata, sia modellando lo sfondo nell'equazione di adattamento. Tuttavia, i tassi di scissione inferiori a questo limite inferiore saranno misurati in modo meno affidabile.
La passivazione superficiale imperfetta può portare a un tethering improprio. Questo porta ad un aumento delle frazioni di perline "bloccate", the che non vanno via durante l'esperimento, o a perline impropriamente legato, che si dissociano dalla superficie molto lentamente. In questo modo viene creata una linea di base più alta ed eventualmente inclinata nei dati elaborati. Si è scoperto che con le foglie di copertura pulite correttamente e la soluzione stock β-caseina appena fatta, questi effetti erano minimi per la maggior parte dei set di dati. Per i set di dati occasionali che mostrano questo, la modifica della funzione di adattamento (per includere una linea di base inclinata) può essere corretta per questo effetto.
Il protocollo attuale può essere esteso in diversi modi. Un ulteriore isolamento dei passaggi meccanicistici dopo l'associazione del sito di destinazione può essere eseguito utilizzando un formato pre-vincolante, in cui la proteina viene iniettata in assenza di cofattori essenziali. Questa idea viene testata iniettando NdeI in assenza di magnesio. In queste condizioni, la proteina si lega al suo sito cognato, ma non lega il DNA. L'iniezione di magnesio dopo questa fase di legame attiva la scissione con conseguente rapida perdita del tallone. L'installazione sperimentale consente anche il controllo della conformazione e della tensione del DNA variando la configurazione del magnete o aggiungendo il flusso. Sotto le forze basse in questi esperimenti, il DNA diventa parzialmente arrotolato. Cambiare leggermente le forze può avere un effetto drammatico sulla conformazione del DNA. Ad esempio, in condizioni di buffer in cui la ricerca target è limitante, variando la conformazione del DNA può verificare l'effetto del salto sulla ricerca di destinazione. In condizioni tampone in cui la fase di idrolisi è limitante al tasso, variando la forza può sondare l'effetto della tensione del DNA sull'idrolisi del legame fosfodiester. Va notato che sotto il basso ingrandimento utilizzato, questi cambiamenti conformazionali non possono essere osservati. I piccoli movimenti risultanti in posizione di tallone devono essere tracciati in ingrandimento più elevato per verificare che la conformazione del DNA possa essere controllata.
L'analisi dei dati può essere estesa in diversi modi. Questo lavoro applica un semplice metodo di conteggio del tallone seguito dal montaggio della curva di una singola funzione esponenziale. Metodi basati sull'analisi del tempo di residenza possono essere utilizzati, cosìcome 20. Le distribuzioni dei tempi di residenza dei singoli DNA possono quindi essere analizzate tramite raccordo a curva o utilizzando tecniche più sofisticate, come il metodo generalizzato dei momenti21.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno conflitti di interesse da divulgare.
Acknowledgments
Questo lavoro è stato sostenuto dalla sovvenzione della National Science Foundation MCB-1715317.
Materials
| Name | Company | Catalog Number | Comments |
| 5 minute Epoxy | Devcon | 14250 | |
| anti-digoxigenin FAB fragments | Roche Diagnostics | 11214667001 | |
| camera and software | Jenoptik | GRYPHAX SUBRA | |
| data analysis software | Vernier Inc. | LP | |
| double sided tape | Grace Biolabs | SA-S-1L | |
| Dulbeccos Phosphate Buffered Saline | Corning | 21-031-CV | |
| ethanol 95% | VWR | 89370-082 | |
| forward primer: digoxigenin-CCAACTTAATCGCCTTGC | Integrated DNA Technologies | n/a | |
| image analysis software | National Institutes of Health | ImageJ | |
| inverted microscope | Nikon | TE2000 | |
| knife printer | Silhouette | ||
| M13mp18 DNA | New England Biolabs | N4040S | |
| MyOne streptavidin beads | Thermo Fisher Scientific | 65601 | |
| NdeI enzyme | New England Biolabs | R0111S | |
| PCR cleanup kit | Qiagen | 28104 | |
| pluronic F-127 | Anatrace | P305 | |
| polyethylene tubing PE-20 | BD Intramedic | 427406 | |
| polyethylene tubing PE-60 | BD Intramedic | 427416 | |
| Q5 Mastermix | New England Biolabs | M0492S | |
| rare earth magnet 0.5" OD 0.25" ID | National Imports | NSN0814 | |
| rare earth magnet 0.75" OD 0.5" ID | National Imports | NSN0615 | |
| reverse primer: biotin-TGACCATTAGATACATTTCGC | Integrated DNA Technologies | n/a | |
| syringe pump | Kent Scientific | Genie Plus | |
| β-Casein from bovine Milk | Sigma-Aldrich | C6905 |
References
- Tock, M. R., Dryden, D. T. The biology of restriction and anti-restriction. Current Opinion in Microbiology. 8 (4), 466-472 (2005).
- Garneau, J. E., et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 468 (7320), 67-71 (2010).
- Pingoud, A., Fuxreiter, M., Pingoud, V., Wende, W. Type II restriction endonucleases: structure and mechanism. Cellular and Molecular Life Sciences. 62 (6), 685-707 (2005).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Sadowski, P. D. Site-specific genetic recombination: hops, flips, and flops. The FASEB Journal. 7 (9), 760-767 (1993).
- Schatz, D. G. Antigen receptor genes and the evolution of a recombinase. Seminars in Immunology. 16 (4), 245-256 (2004).
- Craig, N. L. Tn7: a target site-specific transposon. Molecular Microbiology. 5 (11), 2569-2573 (1991).
- Gori, J. L., et al. Delivery and Specificity of CRISPR/Cas9 Genome Editing Technologies for Human Gene Therapy. Human Gene Therapy. 26 (7), 443-451 (2015).
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics. 11 (9), 636-646 (2010).
- Joung, J. K., Sander, J. D. TALENs: a widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology. 14 (1), 49-55 (2013).
- Alves, J., Urbanke, C., Fliess, A., Maass, G., Pingoud, A. Fluorescence stopped-flow kinetics of the cleavage of synthetic oligodeoxynucleotides by the EcoRI restriction endonuclease. Biochemistry. 28 (19), 7879-7888 (1989).
- Deng, J., Jin, Y., Chen, G., Wang, L. Label-free fluorescent assay for real-time monitoring site-specific DNA cleavage by EcoRI endonuclease. Analyst. 137 (7), 1713-1717 (2012).
- Becker, W. R., et al. High-throughput analysis reveals rules for target RNA binding and cleavage by AGO2. Molecular Cell. 75 (4), 741-755 (2019).
- vanden Broek, B., Lomholt, M. A., Kalisch, S. M., Metzler, R., Wuite, G. J. How DNA coiling enhances target localization by proteins. Proceedings of the National Academy of Sciences. 105 (41), 15738-15742 (2008).
- Gambino, S., et al. A single molecule assay for measuring site-specific DNA cleavage. Analytical Biochemistry. 495, 3-5 (2016).
- Jones, N. D., et al. Retroviral intasomes search for a target DNA by 1D diffusion which rarely results in integration. Nature Communications. 7, 11409 (2016).
- van Aelst, K., et al. Type III restriction enzymes cleave DNA by long-range interaction between sites in both head-to-head and tail-to-tail inverted repeat. Proceedings of the National Academy of Sciences. 107 (20), 9123-9128 (2010).
- Williams, K., et al. A single molecule DNA flow stretching microscope for undergraduates. American Journal of Physics. 79 (11), 1112-1120 (2011).
- Song, D., et al. Tethered particle motion with single DNA molecules. American Journal of Physics. 83 (5), 418-426 (2015).
- Etson, C. M., Todorov, P., Walt, D. R. Elucidating Restriction Endonucleases Reaction Mechanisms via Dwell-Time Distribution Analysis. Biophys Journal. 106 (2), 22 (2014).
- Piatt, S., Price, A. C. Analyzing dwell times with the Generalized Method of Moments. PLoS One. 14 (1), 0197726 (2019).
