

Summary
Se describe un método altamente paralelo para medir el escote específico del sitio del ADN a nivel de molécula única. Este protocolo muestra la técnica mediante la restricción endonucleasa NdeI. El método se puede modificar fácilmente para estudiar cualquier proceso que resulte en una escisión de ADN específica del sitio.
Abstract
La escisión de ADN específica del sitio (SSDC) es un paso clave en muchos procesos celulares, y es crucial para la edición de genes. Este trabajo describe un ensayo cinético capaz de medir SSDC en muchas moléculas de ADN individuales simultáneamente. Los DNA de sustrato amarreado, cada uno de los que contienen una sola copia de la secuencia de destino, se preparan en un canal de flujo microfluídico. Un imán externo aplica una fuerza débil a las perlas paramagnéticas. La integridad de hasta 1.000 DNAs individuales se puede monitorear visualizando las micropersas bajo imágenes de campo oscuro utilizando un objetivo de campo amplio y bajo aumento. La inyección de una restricción endonucleasa, NdeI, inicia la reacción de escote. La microscopía de vídeo se utiliza para registrar el momento exacto de cada escisión de ADN observando el marco en el que el cordón asociado se mueve hacia arriba y fuera del plano focal del objetivo. El recuento de cuentas fotograma a fotograma cuantifica la reacción y un ajuste exponencial determina la velocidad de reacción. Este método permite la recopilación de datos cuantitativos y estadísticamente significativos sobre reacciones SSDC de molécula única en un solo experimento.
Introduction
La escisión de ADN específica del sitio (SSDC) es un paso clave en muchas transacciones genómicas. Por ejemplo, los sistemas de modificación de restricciones bacterianas (RM)1 y CRISPR2 protegen las células del ataque por fagos y plásmidos reconociendo y cortando ADN extraño en secuencias específicas. En el tipo II RM, las endonucleaciones de restricción (RE) reconocen secuencias cortas de 4-8 pares de bases (bp) mediante interacciones entre proteínas y ácidos nucleicos3. Las endonucleasas asociadas a CRISPR, como Cas9, se unen a sitios a través de la hibridación del sitio de destino con crRNAs enlazados a las endonucleasas4. La creación de roturas de doble cadena (DSB) específicas del sitio también son el primer paso en muchos eventos de recombinación de ADN5. Por ejemplo, la diversidad de regiones de enlace de antígenos creadas por la recombinación V(D)J requiere el reconocimiento y la escisión de sitios de destino específicos6. Se sabe que algunos transposones apuntan a secuencias de ADN específicas, así como7. No es de extrañar que muchas nuclelas específicas del sitio implicadas en estos procesos, como Cas9, sean un componente clave de las tecnologías de edición génica8. Además, también se han diseñado nuevas endonucleasas específicas del sitio (es decir, nucleasas de dedos de zinc9 y TALENS10)para editar genomas.
Se han empleado muchos métodos para medir la cinética de la escisión específica del sitio de los ácidos nucleicos. Estos incluyen análisis de gel, fluorescencia11,12, y métodos basados en lasecuenciación 13. Se logró un avance importante con el anclaje de microperlas, lo que permite que los DSB en moléculas individuales de ADN sean detectados por el movimiento de un cordón después de la separación de la hebra. En estos métodos, se emplean diferentes tipos de fuerzas para asegurar la separación de las hebras y el movimiento del cordón después de la escote. En un caso, las trampas ópticas se han utilizado para medir la escisión del ADN mediante EcoRV14. En estos experimentos, la búsqueda de destino es el objetivo de la investigación, con condiciones optimizadas para que el enlace específico del sitio sea el paso de limitación de velocidad. Un inconveniente de las trampas ópticas es que sólo se puede observar un solo ADN a la vez. Además, se debe aplicar una fuerza de tracción grande periódica para probar la separación de la hebra.
Otra técnica utiliza una combinación de flujo y fuerzas magnéticas débiles para tirar del cordón de una manera continua15. De esta manera, se mide la difusión limitada de escote por NdeI. El método empleado permite la medición simultánea de varios cientos de DNA a la vez, lo que permite alcanzar la significancia estadística en un solo experimento. También se han utilizado experimentos basados en pinzas magnéticas. En uno de esos estudios, se estudió una integrasa retroviral mediante la inclusión de un OSD en el oligonucleótido de inserción16. La integración exitosa dio lugar a la incorporación del OSD en el ADN atado y la pérdida del cordón adjunto. En un estudio similar de la restricción de ATP tipo III endonucleasa EcoPI, se observaron decenas de DNA en un solo experimento17. Las pinzas magnéticas tienen la ventaja de que la tensión, así como el bucle de ADN, se pueden controlar y monitorear durante la reacción.
Aquí se presenta un método de molécula única altamente paralelo para medir la cinética SSDC, que aprovecha las mejoras recientes en el anclaje a gran escala de DNAs. Este método es una mejora y extensión de los métodos anteriores utilizados para medir la replicación del ADN18,la longitud del contorno del ADN19,y la escisión por REs15. En esta técnica, los DNA lineales que contienen una sola copia de la secuencia de reconocimiento se preparan con biotina en un extremo y digoxigenina en el otro. La biotina se une a la estreptavidina, que se une covalentemente a un microbead paramagnético. Los complejos de las cuentas de ADN se inyectan en un canal microfluídico que ha sido funcionalizado con fragmentos de la FAB anti-digoxigenina. A continuación, el ADN se une a los puntos de unión superficial mediante la unión de digoxigenina a los fragmentos del FAB adsorbidos. Las fuerzas magnéticas débiles aplicadas con un imán permanente impiden que el cordón se pegue de forma no específica a la superficie. Las muestras se pueden inyectar rápidamente (<30 s) en el canal de flujo para activar la reacción de escote. El flujo se desactiva durante la recopilación de datos. A medida que se corta cada ADN, la hora exacta de la escisión se puede determinar mediante la grabación del marco en el que el cordón se mueve hacia arriba y fuera del plan focal del objetivo, desapareciendo así de la grabación de vídeo. Se puede utilizar un recuento fotograma por fotograma de las perlas restantes para cuantificar el progreso de la reacción.
A continuación se presenta el protocolo completo, así como los datos de ejemplo recopilados con NdeI. Como ejemplo de cómo se puede aplicar la técnica, las tasas de escote para una gama de concentraciones de proteínas se miden en dos concentraciones diferentes de magnesio, un cofactor de metal esencial. Aunque esta aplicación del protocolo utiliza NdeI, el método se puede adaptar para su uso con cualquier nucleasa específica del sitio variando el diseño del sustrato de ADN.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Hacer que la celda de flujo
- Lavado de los cubreobjetos
- Coloque los cubreobjetos en frascos de tinción y sonicar con etanol (EtOH), luego con 1 M KOH (durante 30 minutos cada uno). Para evitar la precipitación de KOH en EtOH, enjuague bien con ddH2O entre lavados.
- Repita el procedimiento tanto EtOH como los pasos de lavado de KOH 1x para un número total de cuatro lavados (dos EtOH y dos KOH). Almacene los cubreobjetos limpios en ddH2O en frascos de tinción.
- Corte los tubos de carga y salida (8 cm de largo) con una maquinilla de afeitar limpia e insértelos en agujeros en un portaobjetos de vidrio limpio. Utilice PE-20 para entrada y PE-60 para toma de corriente. Epoxi durante 5 minutos para asegurar el tubo y recortar cualquier exceso de tubería.
NOTA: Las diapositivas de vidrio miden 2'' x 3'' x 1 mm. Los orificios se perforan en pares separados de 15 mm. Cada par forma cualquier extremo de un solo canal. El tubo con el ID más pequeño se utiliza para la entrada, ya que esto reduce el volumen muerto aguas arriba y, por lo tanto, el tiempo de mezcla requerido (Figura 1). - Alinee y aplique cinta precortado de doble cara con patrón de canal cortado sobre agujeros en la corredera de vidrio. Suaviza con fórceps de plástico para lograr un buen sellado.
NOTA: La cinta de doble cara utilizada en este experimento es de 120 m de espesor, y los canales tienen 2 mm de ancho y 15 mm de largo. Los canales se cortan utilizando una impresorade cuchillos( Tabla de materiales ). Es posible caber hasta cuatro canales en un solo cubreobjetos. Vea el cuadro 1 para una imagen de la célula de flujo. - Después de despegar el respaldo, aplicar un cubreobjetos limpio (secado con aire comprimido) sobre la cinta y suavizar de nuevo con fórceps de plástico para un buen sello.
- Epoxi el borde de la cubierta para sellar la célula de flujo y dejar que se cure.
2. Preparación del ADN etiquetado para el anclaje
- En un tubo de PCR, prepare 50 ml de mezcla de reacción de PCR que contenga 0,02 U/L de ADN polimerasa de alta fidelidad, dNMP de 200 m, imprimación directa de 0,5 m, imprimación inversa de 0,5 m y 250 ng de ADN vectorial M13mp18.
NOTA: Aquí, se eligieron la imprimación delantera (biotina-CCAACTTAATCGCCTTGC) y la imprimación inversa (digoxigenin-TGACCATTAGATACATTTCGC) para amplificar una región de aproximadamente 1.000 bp de largo, que abarca desde las posiciones 6338 a 107 en el genoma circular del ADN M13mp18. Hay un solo sitio NdeI en el centro de la región amplificada. La imprimación delantera está 5ʹ etiquetada con digoxigenina, que une la anti-digoxigenina en el cubreobjetos. La imprimación inversa se 5ʹ etiqueta con biotina, que une las cuentas recubiertas de estreptavidina. - Inserte el tubo PCR en el termociclador y siga el ciclo como se muestra en la Tabla 1.
- Purifique el producto PCR con un kit de limpieza PCR siguiendo el protocolo del fabricante.
NOTA: Utilizando el kit especificado en la Tabla de Materiales,el rendimiento típico del ADN es de 2 g.
3. Tethering de ADN y cuentas
- Preparar 10 mL del tampón A (1 M Tris-HCl [pH a 7,5], 50 mM NaCl, 2 mM MgCl2, 1 mg/ml β-Caseína, 1 mg/ml Pluronic F-127). Degas en un desecador de vacío durante al menos 1 h.
- Para funcionalizar la célula de flujo, inyecte 25 l de fragmentos de FAB anti-digoxigenina (20 g/ml) en PBS en el canal de flujo. Use puntas de carga de gel para encajar en el tubo PE-60. Incubar a temperatura ambiente (RT) durante 30 min.
- Después de la incubación, enjuague el canal tirando de 0,5 ml del tampón A a través del canal utilizando una jeringa. Tenga cuidado de no introducir aire en el canal.
- Después de la funcionalización, monte la celda de flujo en un microscopio invertido. Conecte el tubo de salida a una bomba de jeringa y coloque el tubo de entrada en un tubo de microcentrífuga que contenga el tampón A.
- Tire manualmente de al menos 0,5 ml del búfer A para vaciar el sistema y preparar la bomba. Deje que la bomba funcione a 10 l/min durante al menos 5 minutos para equilibrar el sistema.
- Para preparar las perlas (Tabla de materiales), vórtice la botella de material de perlas y pipetas de 1,6 ml de las perlas de 10 mg/ml en 50 ml de tampón A, a continuación, vórtice.
- Usando un separador magnético, pipetee el búfer y resuspendido en 50 ml del buffer A, luego vórtice.
- Repita el paso 3.7 2x para un total de tres lavados. Para el último lavado, resuspender en 100 l de tampón A y vórtice para lograr una concentración final de 160 g/ml.
- Para complejos el ADN y las perlas, primero prepare 480 l de sustrato de ADN etiquetado con 0,5 pM en el tampón A. A continuación, la pipeta en 20 l de suspensión de cuentas de 160 g/ml, asegurándose de vórtice las perlas antes de pipetear. Colocar en un rotador durante 3 min.
- Después de 3 min, cargue inmediatamente en el canal a un caudal de 10 l/min durante 15 min o hasta que se observe suficiente anclaje del cordón.
NOTA: Las cuentas no deben estar tan densamente embaladas que interactúen entre sí en la superficie (ver sección de discusión). - Para lavar el canal de todas las perlas libres, cambie el tubo de entrada a un tubo fresco de tampón A y fluya a 50 l/min durante al menos 10 minutos o hasta que no se observen perlas sueltas.
4. Recopilación y análisis de datos
- Para preparar la recopilación de datos, coloque el tubo de entrada en un tubo de microcentrífuga que contenga al menos 100 l de NdeI (0,25–4,00 U/ml) en el búfer A. Baje el imán permanente sobre el canal de flujo y coloque el eje de la fuente de luz fuera para la toma de imágenes de campo oscuro.
NOTA: Dos imanes anulares de tierras raras, epoxied juntos, se mantienen 8 mm por encima de la superficie activa del canal de flujo utilizando un poste óptico en voladizo durante la recopilación de datos. Se utiliza una lámpara de cuello de ganso para el curso de luz fuera del eje. - Utilice un microscopio comercial, una cámara de vídeo y un software de recopilación de datos(Tabla de materiales). En el software, haga clic en la pestaña"Exposición"y establezca "Tiempo de exposición"en 10 ms. Haga clic en la pestaña "Timelapse" y establezca "Image Count" en 600, "Duration" a 20 min, y "Interval" en 2 s. Haga clic en "Ejecutar" para iniciar la recopilación de datos.
- En una bomba de jeringa, ajuste el caudal a 150 l/min y el volumen de inyección a 80 l. Pulse"Ejecutar"a 1 min en la recopilación de datos. Después de la inyección, apague la bomba y cierre la válvula para evitar el flujo durante la recolección de datos.
- Una vez recopilados los datos, abra el software de análisis de imágenes (Tabla de materiales). En la pestaña"Archivo",elija "Importar " "Secuencia de imagen". Localice los archivos de imagen en el menú emergente y haga clic en "Abrir".
- Establezca el umbral seleccionando"Ajustar Theshold"en el menú desplegable "Imagen". Utilice la barra deslizante para establecer el valor de umbral para identificar puntos brillantes correspondientes a los cordones de la imagen.
- Cuente los puntos brillantes en cada fotograma haciendo clic en"Analizar partículas"en el menú desplegable"Analizar". Haga clic en "Aceptar", luego elija "Sí" para procesar todas las imágenes. Guarde el archivo de resultados.
NOTA: Esto guardará un archivo de datos que contiene el número de perlas en cada fotograma de vídeo grabado. - Abra el software de análisis de datos (Tabla de materiales) e importe el archivo de resultados haciendo clic en " Importar desde archivo detexto" en el menú desplegable "Archivo". Trazar los datos de recuento de cuentas frente a la hora.
- Ajuste el número de cuentas frente al tiempo haciendo clic en "Ajuste de curva" en el menú desplegable"Analizar". Elija la ecuación "Exponente Natural" y haga clic en "Probar Ajuste " "OK".
NOTA: Solo deben incluirse en la región de montaje los datos registrados después de la inyección de la muestra. El parámetro de ajuste en el exponente de la función de ajuste será la tasa de escisión.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Usando esta técnica, las tasas SSDC de NdeI se midieron para una gama de concentraciones de proteínas (0,25–4,00 U/ml) en dos concentraciones diferentes de magnesio (2 mM y 4 mM). Cada una de estas condiciones se replicó al menos dos veces, con unos pocos cientos a 1.000 DNAs atados por experimento. La Figura 2 describe el diseño experimental. En la figura 3 se muestran ejemplos de detalles de recopilación y análisis de datos. La Figura 4 ilustra cómo la tasa depende de la concentración de proteínas en las dos concentraciones de magnesio. Se puede observar que a concentraciones de proteínas suficientemente bajas, la tasa es proporcional a la proteína e independiente del magnesio. Para concentraciones de proteínas suficientemente altas, la tasa depende del magnesio pero es independiente de la concentración de proteínas.
| Paso | Descripción | Temperatura (oC) | Hora (s) |
| 1 | Desnaturalización | 98 | 30 |
| 2 | Derretir | 98 | 10 |
| 3 | Recueza | 60 | 30 |
| 4 | Extender | 72 | 30 |
| 5 | Extensión final | 72 | 120 |
Tabla 1: Parámetros PCR. Se muestran las temperaturas y duraciones de los pasos del programa termociclor utilizados en el paso 2.2 del protocolo. Los pasos de fusión, recocido y extensión (pasos 2, 3 y 4) se repiten 30 veces.
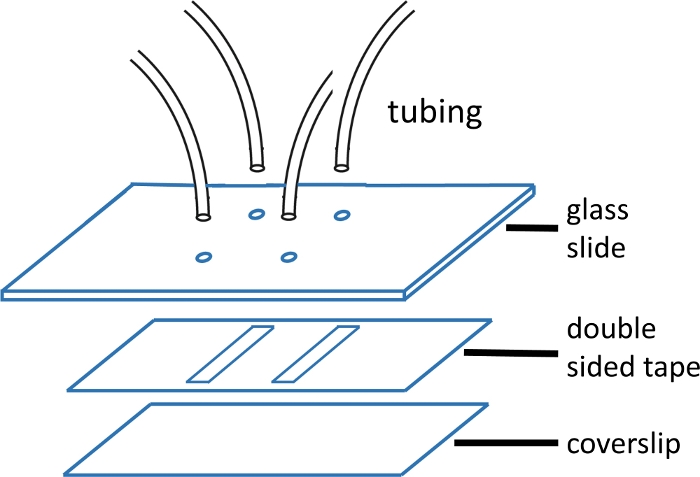
Figura 1: Construcción de celdas de flujo microfluídicas.
La corredera de vidrio superior (2'' x 3'', 1 mm de espesor) está preperforada con orificios que coinciden con el patrón de canal. Los tubos de entrada y salida se insertan en los orificios y se fijan con epoxi antes de fijar la cinta y el vidrio de la cubierta. La cinta de doble cara está precortado con patrón de canal. La diapositiva inferior (#1 o #1.5 de cristal de cubierta) se limpia previamente utilizando el protocolo descrito en el texto principal. Una vez montado, el borde del vidrio de la cubierta se sella con epoxi. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Diseño experimental.
(A) Método de anclaje de ADN. El ADN de doble cadena (1 kbp) etiquetado con digoxigenina en el extremo 5ʹ se une a la superficie de la célula de flujo a través de la interacción antidigoxigenina-digoxigenina. El extremo 3ʹ del ADN, etiquetado con biotina, se une a un micropera mediante la interacción estreptavidina-biotina. El sitio de escote NdeI se encuentra en el centro del ADN. (B) Configuración experimental durante la recopilación de datos que muestra el imán y la posición objetiva. El imán permanente mantiene una fuerza ascendente débil en el cordón durante la reacción de escote. Haga clic aquí para ver una versión más grande de esta figura.
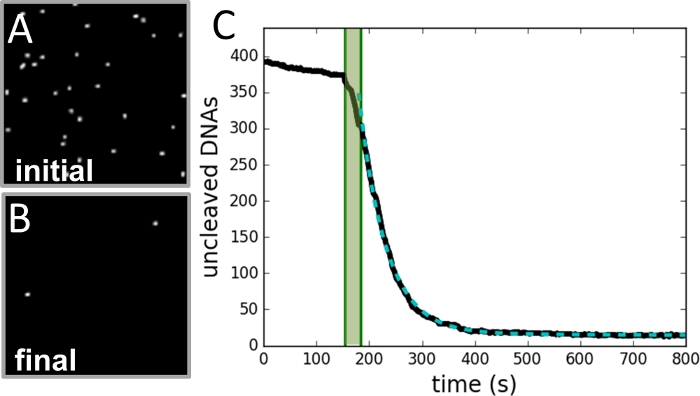
Figura 3: Ejemplo de recopilación y análisis de datos.
(A) Imagen de la región de las cuentas tomadas antes de iniciar la reacción del escote. (B) Imagen de la misma región una vez completada la reacción. (C) Trazado del número de cuentas frente al tiempo (curva negra) según se determine a partir de cada fotograma del registro de vídeo. El área verde sombreada marca el período de inyección de enzima y no está incluida en el ajuste. Los datos se ajustan a una sola curva exponencial (curva verde discontinua), con una constante de decaimiento igual a la tasa de reacción. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: La escisión NdeI depende de las concentraciones de proteína y magnesio.
Gráfica de la tasa SSDC medida de NdeI para un rango de concentraciones de proteínas en dos concentraciones diferentes de magnesio: 2 mM (círculos azules) y 4 mM (cuadrados verdes). Las barras de error representan SEM. Las curvas discontinuas son líneas de tendencia y no representan ajustes a la teoría. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
El protocolo se puede utilizar para medir la cinética de cualquier sistema SSDC, siempre que se observe la separación de hebras durante el experimento. La detección de escote se ve afectada por la observación del desprendimiento del cordón amarreado y, por lo tanto, marca el instante de separación de la hebra. Todos los pasos anteriores se producen antes de la detección del escote; por lo tanto, sólo se registra el tiempo total de tránsito.
El cubreobjetos de células de flujo se funcionaliza mediante adsorción no específica de proteína de anticuerpos al vidrio limpio. El vidrio insuficientemente limpio puede afectar la unión del anticuerpo. En el anclaje, la densidad del cordón debe ser lo suficientemente baja para que las perlas no interactúen. La densidad superficial de los puntos de unión se puede controlar mediante la concentración de anticuerpos durante la funcionalización. El número total de cuentas depende del tamaño del campo de visión. En este caso, unos pocos cientos a 1.000 cuentas atadas eran suficientes para buenas estadísticas y evitaban interacciones de perlas. Durante la inyección de cuentas, el anclaje de la superficie fue monitoreado a través de video en vivo. La inyección de cuentas se detuvo cuando el recuento estimado de cuentas fue de entre 500 y 1.000 perlas.
Las tasas de escisión más rápidas que se pueden medir con precisión están limitadas por el tiempo de mezcla de la celda de flujo. El tiempo de mezcla en las células de flujo laminar está influenciado por varios factores. La difusión a la superficie es un paso clave; por lo tanto, el tiempo de mezcla depende del coeficiente de difusión del reactivo. La cizalladura significativa que se produce en el tubo de entrada, que transporta la muestra al canal de flujo desde el depósito de la muestra, puede aumentar el tiempo necesario para garantizar una mezcla adecuada en la superficie de reacción en el canal. Se encontró que el tiempo de mezcla podría reducirse reduciendo el volumen muerto aguas arriba y aumentando el caudal. Con un diámetro interior de 380 m y una longitud máxima de 8 cm para el tubo de entrada (y un caudal de 150 l/min), se encontró que el tiempo de inyección podía reducirse a 20 s sin afectar a la tasa de escisión medida. Dado que el tiempo de mezcla depende del coeficiente de difusión del reactivo, debe determinarse por separado para cada enzima o activador de escote estudiado.
El método de anclaje permite la ruptura no específica de la teadura, presumiblemente debido a la disociación del complejo digoxigenina-anticuerpo o a la liberación del anticuerpo de la superficie. Esto da como resultado una tasa de pérdida de cordón de fondo reproducible presente antes de la inyección de enzima de 3 x 10-4 s-1. Este efecto sistemático se puede corregir ya sea restando la tasa de fondo de la tasa de escisión medida, o modelando el fondo en la ecuación de ajuste. Sin embargo, las tasas de escote inferiores a este límite inferior se medirán de forma menos fiable.
La pasivación de la superficie imperfecta puede conducir a un anclaje inadecuado. Esto conduce al aumento de las fracciones de cuentas "pegadas", que no desaparecen durante el experimento, o a cuentas mal atadas, que se disoccian de la superficie muy lentamente. Esto crea una línea base más alta y posiblemente inclinada en los datos procesados. Se encontró que con los resbalones de cubierta correctamente limpiados y la solución de stock de β-casein recién hecha, estos efectos eran mínimos para la mayoría de los conjuntos de datos. Para los conjuntos de datos ocasionales que muestran esto, la modificación de la función de ajuste (para incluir una línea base inclinada) puede corregir este efecto.
El protocolo actual se puede ampliar de varias maneras. El aislamiento adicional de los pasos mecanicistas después de la unión al sitio de destino se puede realizar utilizando un formato de prea vinculante, en el que la proteína se inyecta en ausencia de cofactores esenciales. Esta idea se prueba inyectando NdeI en ausencia de magnesio. En estas condiciones, la proteína se une a su sitio cognado, pero no corta el ADN. La inyección de magnesio después de este paso de unión activa el escote dando como resultado una pérdida rápida del cordón. La configuración experimental también permite el control de la conformación y tensión del ADN variando la configuración del imán o añadiendo flujo. Bajo las fuerzas bajas en estos experimentos, el ADN se enrolla parcialmente. Cambiar ligeramente las fuerzas puede tener un efecto dramático en la conformación del ADN. Por ejemplo, en condiciones de búfer en las que la búsqueda de destino es la limitación de velocidad, variar la conformación del ADN puede probar el efecto de saltar en la búsqueda de destino. En condiciones de amortiguación en las que el paso de hidrólisis es limitador de velocidad, la variación de la fuerza puede sondear el efecto de la tensión del ADN en la hidrólisis de unión de fosfodiester. Cabe señalar que bajo el bajo aumento utilizado, estos cambios conformales no pueden ser observados. Los pequeños movimientos resultantes en la posición del cordón tienen que ser rastreados bajo mayor aumento para verificar que la conformación del ADN puede ser controlada.
El análisis de datos se puede ampliar de varias maneras. Este trabajo aplica un método de recuento de cordones simple seguido de ajuste de curva de una sola función exponencial. Se pueden utilizar métodos basados en el análisis del tiempo de residencia, asícomo 20. Las distribuciones de los tiempos de residencia de los DNAs individuales se pueden analizar mediante el ajuste de curvas o mediante técnicas más sofisticadas, como el método generalizado de los momentos21.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen conflictos de intereses que revelar.
Acknowledgments
Este trabajo fue apoyado por la subvención MCB-1715317 de la National Science Foundation.
Materials
| Name | Company | Catalog Number | Comments |
| 5 minute Epoxy | Devcon | 14250 | |
| anti-digoxigenin FAB fragments | Roche Diagnostics | 11214667001 | |
| camera and software | Jenoptik | GRYPHAX SUBRA | |
| data analysis software | Vernier Inc. | LP | |
| double sided tape | Grace Biolabs | SA-S-1L | |
| Dulbeccos Phosphate Buffered Saline | Corning | 21-031-CV | |
| ethanol 95% | VWR | 89370-082 | |
| forward primer: digoxigenin-CCAACTTAATCGCCTTGC | Integrated DNA Technologies | n/a | |
| image analysis software | National Institutes of Health | ImageJ | |
| inverted microscope | Nikon | TE2000 | |
| knife printer | Silhouette | ||
| M13mp18 DNA | New England Biolabs | N4040S | |
| MyOne streptavidin beads | Thermo Fisher Scientific | 65601 | |
| NdeI enzyme | New England Biolabs | R0111S | |
| PCR cleanup kit | Qiagen | 28104 | |
| pluronic F-127 | Anatrace | P305 | |
| polyethylene tubing PE-20 | BD Intramedic | 427406 | |
| polyethylene tubing PE-60 | BD Intramedic | 427416 | |
| Q5 Mastermix | New England Biolabs | M0492S | |
| rare earth magnet 0.5" OD 0.25" ID | National Imports | NSN0814 | |
| rare earth magnet 0.75" OD 0.5" ID | National Imports | NSN0615 | |
| reverse primer: biotin-TGACCATTAGATACATTTCGC | Integrated DNA Technologies | n/a | |
| syringe pump | Kent Scientific | Genie Plus | |
| β-Casein from bovine Milk | Sigma-Aldrich | C6905 |
References
- Tock, M. R., Dryden, D. T. The biology of restriction and anti-restriction. Current Opinion in Microbiology. 8 (4), 466-472 (2005).
- Garneau, J. E., et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 468 (7320), 67-71 (2010).
- Pingoud, A., Fuxreiter, M., Pingoud, V., Wende, W. Type II restriction endonucleases: structure and mechanism. Cellular and Molecular Life Sciences. 62 (6), 685-707 (2005).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Sadowski, P. D. Site-specific genetic recombination: hops, flips, and flops. The FASEB Journal. 7 (9), 760-767 (1993).
- Schatz, D. G. Antigen receptor genes and the evolution of a recombinase. Seminars in Immunology. 16 (4), 245-256 (2004).
- Craig, N. L. Tn7: a target site-specific transposon. Molecular Microbiology. 5 (11), 2569-2573 (1991).
- Gori, J. L., et al. Delivery and Specificity of CRISPR/Cas9 Genome Editing Technologies for Human Gene Therapy. Human Gene Therapy. 26 (7), 443-451 (2015).
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics. 11 (9), 636-646 (2010).
- Joung, J. K., Sander, J. D. TALENs: a widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology. 14 (1), 49-55 (2013).
- Alves, J., Urbanke, C., Fliess, A., Maass, G., Pingoud, A. Fluorescence stopped-flow kinetics of the cleavage of synthetic oligodeoxynucleotides by the EcoRI restriction endonuclease. Biochemistry. 28 (19), 7879-7888 (1989).
- Deng, J., Jin, Y., Chen, G., Wang, L. Label-free fluorescent assay for real-time monitoring site-specific DNA cleavage by EcoRI endonuclease. Analyst. 137 (7), 1713-1717 (2012).
- Becker, W. R., et al. High-throughput analysis reveals rules for target RNA binding and cleavage by AGO2. Molecular Cell. 75 (4), 741-755 (2019).
- vanden Broek, B., Lomholt, M. A., Kalisch, S. M., Metzler, R., Wuite, G. J. How DNA coiling enhances target localization by proteins. Proceedings of the National Academy of Sciences. 105 (41), 15738-15742 (2008).
- Gambino, S., et al. A single molecule assay for measuring site-specific DNA cleavage. Analytical Biochemistry. 495, 3-5 (2016).
- Jones, N. D., et al. Retroviral intasomes search for a target DNA by 1D diffusion which rarely results in integration. Nature Communications. 7, 11409 (2016).
- van Aelst, K., et al. Type III restriction enzymes cleave DNA by long-range interaction between sites in both head-to-head and tail-to-tail inverted repeat. Proceedings of the National Academy of Sciences. 107 (20), 9123-9128 (2010).
- Williams, K., et al. A single molecule DNA flow stretching microscope for undergraduates. American Journal of Physics. 79 (11), 1112-1120 (2011).
- Song, D., et al. Tethered particle motion with single DNA molecules. American Journal of Physics. 83 (5), 418-426 (2015).
- Etson, C. M., Todorov, P., Walt, D. R. Elucidating Restriction Endonucleases Reaction Mechanisms via Dwell-Time Distribution Analysis. Biophys Journal. 106 (2), 22 (2014).
- Piatt, S., Price, A. C. Analyzing dwell times with the Generalized Method of Moments. PLoS One. 14 (1), 0197726 (2019).
